
The New Mellenium 2000


The New Millennium
The main scientific research efforts were directed towards better understanding the function and control of CFTR and continuing efforts to treat the basic genetic defect with either gene replacement therapy or pharmacological correctors or potentiators of the mutant gene.
In 1999 three research teams in London, Oxford and Edinburgh were encouraged to form the UK Gene Therapy Consortium (GTC) and eventually a multidose gene therapy trial started in 2013 and successfully completed in 2015; unfortunately the modest effect was not sufficient to take the product forward (www.cfgenetherapy.org.uk)
The use of CF animal models remained an essential part of research, particularly CF mice, which had been available since the early Nineties (Snouweart et al, 1992; Dorin et al, 1992; Radcliff et al, 1993). Eventually towards the end of the decade a CF pig was produced (Rodgers et al, 2008) and also a CF ferret (Sun X et al, 2010).
In N. America, and to a lesser extent in mainland Europe, the emphasis of research has been on a variety of pharmacological correctors and potentiators of the abnormal gene (www. cff.org/treatments/pipeline) Also in Israel the use of gentamicin for “stop mutations” (Wilschanski et al, 2000) and subsequently of PTC 124 (Ataluren) (Kerem et al, 2008) were under investigation.
CFTR modulator drugs to treat specific genetic mutations were introduced in the second decade. The first, ivacaftor, became available in 2012 (Ramsey BW et al, 2011) and undoubtedly represented the most important advance up to that time. This and subsequent modulator drugs introduce during the second decade radically changed the outlook for the majority of people with cystic fibrosis – who lived in countries who could afford the new drugs. From 2013 a series of modulators for treating other mutations, including the most frequent Phe508del, were undergoing clinical trials either alone of with Ivacaftor (Keating et al, 2018).
The most recent combination of elexacaftor/tezacaftor/ivacaftor ((known as Trikafta in the USA, Kaftrio in Europe) represented a really major unprecedented advance which has radically changed the outlook for over 90% of patients who have one or two Phe508del mutations; the results were quite dramatic. Undoubtedly, the treatment of people with CF had entered a new era (Keating et al, 2018; Middleton et al, 2019; Heijerman et al,2019). Unfortunately, Trikafta/Kaftrio is very expensive and were unaffordable in some countries.
Early diagnosis in the first weeks by neonatal CF screening, early expert advice, support and monitoring by a specialist CF team, and early appropriate treatment of respiratory infection and malabsorption are now well established as essential and should be the right of all people with CF both now and in the future. Chronic gastrointestinal problems of pain and distal intestinal obstruction syndrome are still relatively common and it is encouraging that they are receiving more attention. A greater proportion of care will be provided from Specialist CF Centres – virtually all advances in clinical care have occurred at such centres. The persisting inequalities of care so clearly revealed in the past and yet still present as revealed by the increasingly valuable CF registries in North America, the UK and elsewhere, hopefully will lessen.
More lung transplants will be required and measures are being taken to increase the supply of donor organs where this is a problem. Other major and complex problems increased as the population ages e.g. diabetes, liver disease, osteoporosis, pregnancy, infertility, chronic renal problems, drug allergies as well as complex psychosocial issues. There was increasing use of home care and CF Nurse Specialists as part of the CF centre team. Protocols for prevention of cross-infection were even more important as new organisms emerge and resistance and allergies to current antibiotics increased.
Carrier testing of relatives, the option of antenatal screening and, if positive diagnosis, and pre-implantation diagnosis (PIGD) for high risk couples should be encouraged and available. Unfortunately, this opportunity to ensure an infant of two carriers does not have CF by the use of PIGD does not receive the attention it deserves at present.
Major efforts to treat the basic defect evermore efficiently will continue. It is very likely that either gene replacement, into which there is still very active research, or even more impressive pharmacological treatments with the new modulators or both will effectively normalise, or significantly further improve, the disturbed physicochemical condition within the CF airways and other organs, so that much less treatment or even no other treatment will be required for the respiratory tract.
There has never been a time when there was more hope of major progress in CF care than the present. In contrast with the really hopeless outlook in the early years, the present state of knowledge, clinical care and the involvement of so many highly regarded scientists would have been beyond belief even 25 years ago. In fairness to the early workers, there have also been massive advances in medicine, science and technology generally, many of which have facilitated the advances in CF research.
While research, prevention, improvement in diagnosis, treatment and provision of optimal care for people at all stages of the condition will remain of the highest priority, progress both in the pharmacological treatment and gene replacement therapy to correct the basic defect is definitely gaining momentum and success in this area is likely to benefit all people with CF even patients whose condition is more advanced and those with unusual mutations..
2000
Andersson C, Roomans GM. Activation of delta F508 CFTR in a cystic fibrosis respiratory epithelium cell line by 4-phenylbutyrate, genistein and CPX. Eur Respir J 2000; 15:937-941. [PubMed]
4-phenylbutyrate allows the DF508 CFTR to escape degradation in the body of the cell so it can to be transported to the cell surface. Genistein and 8-cyclopentyl-1, 3-dipropylxanthine (CPX) act by stimulating chloride ion efflux from the cell by increasing the probability of the CFTR channel being open. The authors suggested that a combination of 4-phenylbutyrate and genistein may be useful in a potential pharmacological therapy for people with CF with the deltaF508 mutation.
– Considerable attention has been given to the effects of these three compounds on the functioning of DF508 CFTR. During the decade major developments in drug therapy to modify or correct the basic genetic defect became the main research effort in N. America and gene replacement therapy, led by the UK Gene Therapy Consortium, would be the main research focus in the UK.
Bhura-Bandali FN, Suh M, Man SF,Clandinin MT. The deltaF508 mutation in the cystic fibrosis transmembrane conductance regulator alters control of essential fatty acid utilization in epithelial cells. J Nutr 2000; 130:2870-2875. [PubMed]
Essential fatty acid (EFA) incorporation into phospholipid is influenced by chloride channels, suggesting that CFTR may exert a regulatory effect on EFA metabolism. The authors state that the observations in this paper suggest that CF results in a defect in the utilization of 18:2(n-6), which they attribute in part to the defective CFTR.
– This is yet another twist to the EFA story in cystic fibrosis. Recently Freedman et al (1999 above) had shown a membrane lipid imbalance in the ileum, pancreas, and lung from cftr (-/-) mice characterized by an increase in phospholipid-bound arachidonic acid and a decrease in phospholipid-bound docosahexaenoic acid. This finding caused considerable interest at the 1999 North American CF Conference but unfortunately subsequent studies failed to confirm the fundamental importance of the findings. A later report from Freedman and colleagues in 2007 suggested that DHA therapy may release endogenous inhibitors of inflammation although the initial enthusiasm has waned (Freedman SD et al, 2004; Beharry et al, 2007 – both below). The subject of fatty acids in CF was still an area of active research ion 2023.
Bombieri C, Luisetti M, Belpinati F, Zuliani E, Beretta A, Baccheschi J, Casali L, Pignatti PF. Increased frequency of CFTR gene mutations in sarcoidosis: a case/control association study. Eur J Hum Genet 2000; 8:717-720.[PubMed]
A complete screening of the CFTR gene by DGGE and DNA sequencing was performed in patients with sarcoidosis. In 8/26 cases, missense and splicing CFTR gene mutations were found, a significant difference over controls (9/89) from the same population. A trend towards disease progression was observed in patients with CFTR gene mutations compared to patients without mutations.
These data suggest that CFTR gene mutations predispose to the development of sarcoidosis.
Cade A, Walters MP, McGinley N, Firth J, Brownlee KG, Conway SP, Littlewood JM. Evaluation of fecal pancreatic elastase-1 as a measure of pancreatic exocrine function in children with cystic fibrosis. Pediatr Pulmonol 2000; 29:172-176. [PubMed]

Fig 1. Alan Cade. author’s photo
Experience of pancreatic elastase-1 measurements (fecal EL-1) from a large series of 142 patients from the paediatric CF centre in Leeds. The value in 135 pancreatic insufficient (PI) patients was 10 microg/g stool (2.5-33); of the 7 pancreatic sufficient (PS) patients, 698 microg/g stool (400.5 – 824.5), and that of the non-CF controls, 615 microg/g stool (420-773). There is an impressive clear cut separation of the groups.
This is one of a number of papers confirming the value of a relatively new and reliable non-invasive test of pancreatic function using a single small faecal specimen; also the result is still valid even when patient is taking pancreatic enzyme replacement therapy. Fecal EL-1 is now used as evidence of pancreatic insufficiency in screened CF infants and is reliable over the age of 2 weeks; in older CF patients at diagnosis; for confirming the need for pancreatic enzymes in patients referred to the CF centre from other hospitals who are already taking enzymes (experience shows that some are pancreatic sufficient and do not require the enzyme supplements they have been prescribed); for annual monitoring of PS patients to detect the onset of PI; and as supporting evidence when excluding the diagnosis of CF in patients attending the pediatric gastroenterology clinic for investigation of chronic diarrhoea.
The test was undoubtedly a very valuable addition to the tests for pancreatic insufficiency and one of the most useful of any previously described. The first brief report of EL-1 in CF children was in 1997 from Great Ormond Street, London (Wallis et al. Lancet 1997; 350:1001.above [PubMed]) the test having been described earlier in adults by Loser et al. (Gut 1996; 39:580-586. [PubMed] and others.
Dr Alan Cade (figure 1) was working in the Leeds Regional CF Centre at the time of this study and is now Consultant Paediatrician in Plymouth UK where he is Consultant Paediatrician and Director of the Paediatric CF unit.
Cheng K, Smyth RL, Motley J, O’Hea U, Ashby D. Randomized controlled trials in cystic fibrosis (1966-1997) categorized by time, design, and intervention. Pediatr Pulmonol 2000; 29:1-7.[PubMed]
Ros Smyth, Deborah Ashby and colleagues developed a register of randomized controlled trials (RCTs) in CF and have studied when they were performed, their design, and what interventions were investigated. They identified 506 RCTs; 37.5% were identified solely as abstract reports in conference proceedings. There has been about a 30-fold increase in the number of RCTs in CF since 1966. A high proportion of the RCTs (72.7%) had a sample size of 30 or less, and only 8.7% were multicenter trials. Reporting of study design was poor: in 51.4% the report did not state whether there was any blinding in the trial design; 53.6% of studies were of crossover design. The most common interventions studied were antibiotic treatments and physiotherapy, but a number of commonly used therapies had been evaluated only in a small number of patients.
Although the number of RCTs of interventions in CF patients has increased over the last 25 years, the sample sizes of these trials are generally too small to indicate whether the intervention was effective, and very few were multicenter.
Future RCTs in CF are more likely to provide clinically useful answers if higher numbers of patients are recruited into large, well-designed multicenter trials. This should be a priority of the organization of future research in CF. This is a very important point particularly if interventions, which appear obviously beneficial and subsequently prove to be so, are delayed for years for want of a clinical trial that reaches rigorous Cochrane standards. This has occurred in the case of inhaled anti-pseudomonal antibiotics both for early eradication and for stabilisation of chronic infection.
Conway SP, Morton AM, Oldroyd B, Truscott JG, White H, Smith AH, Haigh I. Osteoporosis and osteopenia in adults and adolescents with cystic fibrosis: prevalence and associated factors. Thorax 2000; 55:798-804. [PubMed] Sixty six percent of 114 patients attending the Leeds Regional CF Centre had osteopenia or osteoporosis. The clinical score correlated significantly with bone mineral density (BMD) at the lumbar spine and femoral neck, and with total body BMD. Oral steroid use was significantly associated with reduced BMD at the lumbar spine and femoral neck.
Following the first extensive report by Aris et al (1998 above), there was increasing interest in osteoporosis as more people with CF reached adulthood and many of them had osteoporosis. This present study and others confirmed that osteopenia and osteoporosis were common findings in adults with CF, particularly those with severe disease and particularly those who had used significant amounts of corticosteroids.
Mrs Alison Morton (fig. 2) is the senior dietitian at the Leeds Regional Adult CF Unit and closely involved in many publications from that unit and also she is on the expert advisory committees of the UK CF Trust.

Fig. 2: Alison Morton
Conway SP, Etherington C, Munday J, Goldman MH, Strong JJ, Wootton M. Safety and tolerability of bolus intravenous colistin in acute respiratory exacerbations in adults with cystic fibrosis. Ann Pharmacother 2000; 34:1238-1242. [PubMed]
This study confirmed that an intravenous bolus of 2 mega-units of colistin in 10 ml of normal saline was safe and tolerated by patients with indwelling venous access systems.
An important and practically useful paper as intravenous colistin was increasingly used as bacterial resistance to other antibiotics became more common; also previously IV colistin had been considered to be associated with unacceptable side effects. Previously it had been recommended that colistin should be given as a slow infusion when given through peripheral veins to avoid thrombophlebitis. The present study describes tolerability and use of indwelling venous access systems rather than peripheral veins.
Cooper RA, Wigley P, Burton NF. Susceptibility of multiresistant strains of Burkholderia cepacia to honey. Lett Appl Microbiol 2000; 31:20-24. [PubMed]
Twenty strains of Burkholderia cepacia, isolated principally from the sputum of people with CF, were tested in the laboratory for their susceptibility to eight antibiotics. All strains exhibited multiple, but not identical, patterns of antibiotic resistance. However, all strains exhibited susceptibility to concentrations of honey below 6% (v/v). The authors suggested that honey may have a potential role in the clinical management of B. cepacia infections.
As yet (2023) honey has not proved to be of value in the treatment of people with CF although it has been used with success in other infections particularly involving the skin. Professor John Govan of Edinburgh had a PhD student and a chemist working on the anti-bacterial effect of honey but they were unable to identify the active component. One experienced CF physician even tried treating an adult patient with CF with nebulised solution of honey but again without obvious clinical benefit.
Döring G, Conway SP, Heijerman HGH, Hodson ME, Hoiby N, Smyth A, Touw DJ, for the Consensus Committee. Antibiotic therapy against Pseudomonas aeruginosa in cystic fibrosis: a European Consensus. Eur Respir J 2000; 16:749-767. [PubMed]

Fig 2a Gerd Döring
This document was the result of one of the valuable consensus meetings organised at Artimino in northern Italy by Prof. Gerd Döring and the European CF Society of which he was President. In 2006 Gerd Döring later became editor of the Journal of Cystic Fibrosis. In this publication there were consensus answers to 24 important questions based on current evidence by a panel of 34 European CF experts. (more details in the PubMed abstract).
The UK CF Trust produced a consensus document on antibiotics in 2002 – Antibiotic Treatment for Cystic Fibrosis. 2002. Full text of the extensively revised 2009 third edition is now available on the CF Trust website (www.cysticfibrosis.org.uk).
Gerd Döring (Fig 2a) (1948- 2013) Gerd was a world authority on infection and inflammation in the CF lung and made major contributions over his lifetime to the understanding of the pathophysiology of cystic fibrosis Gerd has also given outstanding leadership to the European Cystic Fibrosis Society. During his time as President, from 1998 to 2006, he was instrumental in moving the society from a working group to a vibrant scientific society. Gerd was the Editor-In-Chief of the Journal of Cystic Fibrosis since 2006 and was due to retire from his role at the end of 2013.
Dudding T, Wilcken B, Burgess B, Hambly J, Turner G. Reproductive decisions after neonatal screening identifies cystic fibrosis. Arch Dis Child Fetal 2000; 82:F124-127. [PubMed]
The extensive New South Wales experience of neonatal CF screening from 1981 to 1996 indicated that two thirds of women who had a CF infant chose to avoid having another child with CF. In subsequent pregnancies 66% had antenatal diagnosis of whom 69% terminated or would have terminated had the fetus been affected. The 59% who decided against further pregnancies did so to avoid having a further child with CF.
As was also shown in East Anglia UK and Brittany France the presence of a neonatal CF screening programme appears to have an overall effect of reducing the future incidence of CF in the newborns in that region. A similar effect was shown following the introduction of antenatal screening in Edinburgh (Cunningham S, Marshall T. Arch Dis Child 1998; 78:345-348. above [PubMed]). Similar findings re. antenatal diagnosis and termination were reported in the large neonatal CF screening programme from Brittany (Scotet et al, 2000 above [PubMed]). However, it may be that with the steadily improving prognosis termination will become increasingly unacceptable to potential parents. This was said to be one factor in discontinuing the successful antenatal CF screening in Edinburgh.
Edenborough FP, Mackenzie WE, Stableforth DE. The outcome of 72 pregnancies in 55 women with cystic fibrosis in the United Kingdom 1977-1996. Brit J Obstet Gynaec 2000; 107:254-261. [PubMed]

Fig 3. Frank Edenborough
Frank Edenborough (fig, 3), Director of the Adult CF Centre in Sheffield, reviews 72 pregnancies in women with CF in the UK, the outcomes were known for 69; there were 48 live births (70%) of which 22 were premature (46%); 14 therapeutic abortions (20%); and 7 miscarriages (10%). There were no stillbirths, neonatal or early maternal deaths. Three major fetal anomalies were seen, but no infant had cystic fibrosis. The outcomes for the infants of women with CF are generally good but are variable for the mother – lung function being the most significant determining factor. All pregnancies should be planned with prior counselling and monitored by dedicated cystic fibrosis teams, including obstetricians who are experienced in managing high risk pregnancies (also Siegel & Siegel, 1960 above; Cohen et al, 1980 above; Gilljam et al, 2000 below). There are more recent European guidelines – Edenborough et al. Guidelines for management of pregnancy in women with cystic fibrosis. J Cyst Fibros 2008; Suppl 1:S1-32 [PubMed])
Elborn JS, Prescott RJ, Stack BH, Goodchild MC, Bates J, Pantin C. Ali N, Shale DJ, Crane M. Elective versus symptomatic antibiotic treatment in cystic fibrosis patients with chronic Pseudomonas infection of the lungs. Thorax 2000; 55:355-358.[PubMed]
A clinical report from Copenhagen had suggested that regular 3-monthly courses of IV antibiotics increased survival of patients with CF who were chronically infected with Pseudomonas species (Szaff et al. 1983 above). In this present UK multicentre study 60 patients with CF, chronically infected with P. aeruginosa, were randomised to the two treatment arms (elective or symptomatic) and followed clinically at annual reviews. Unfortunately the patients in the symptomatic group (IV only if required for clinical condition) received a mean of three antibiotic treatments each year, very similar to those in the elective group who received four antibiotic treatments during each year of the study.
The study failed to demonstrate an advantage of a policy of elective 3-monthly antibiotic treatment over symptomatic treatment in patients with CF chronically infected with Pseudomonas species. Unfortunately not an entirely satisfactory study as B.cepacia deaths complicated the results; also the symptomatic group received almost as many courses of IV antibiotics as the treatment group – no less than 3 courses of IV antibiotic in the year compared with 4 in the elective group. So the question is still not finally answered as to whether regular 3-monthly IV treatment, irrespective of clinical state, should be given to patients with chronic P. aeruginosa infection. A very important difference to note between the two studies (UK and Copenhagen) is the patients were receiving regular nebulised antibiotics in this UK study but these were not used by chronically infected patients at the time of the original Danish report (Szaff et al. 1983 above).
In recent years (2014) many UK CF centres have a very low threshold for treating with IV antibiotics and many patients with chronic P. aeruginosa infection are actually receiving almost 3-monthly courses of IV antibiotics, being treated if they show even minimal signs of deterioration. It is now, and quite correctly, a matter of clinical judgement in most CF centres – if the chronically infected patient shows slow deterioration of respiratory function they receive 3 monthly IV antibiotics, if they are absolutely stable the intervals between the courses are increased. A few remain totally stable with inhaled antibiotics, which all chronically infected patients should be receiving, and may go for many years without the need for IV antibiotics.

Fig. 4: Stuart Elborn CBE
Professor Stuart Elborn (fig. 4) at this time is Director of the Adult CF Centre at the Royal Brompton Hospital London and previously the Professor of Respiratory Medicine in Queens University Belfast. At this time he was Chairman of the CF Trust’s Research Advisory Committee and President of the European CF Society. He is one of Europe’s leading authorities on cystic fibrosis. Stuart was awarded the CBE in 2013 for his services to medicine.
Feigelson J, Pecau Y, Poquet M, Terdjman P, Carrere J, Chazalette JP, Ferec C. Imaging changes in the pancreas in cystic fibrosis: a retrospective evaluation of 55 cases seen over a period of 9 years. J Pediatr Gastroenterol Nutr 2000; 30:145-151. [PubMed]
The aim of this study was to follow-up the evolution of the CF pancreatic changes in 55 people with CF to define its successive stages in correlation with the clinical, biochemical, and imaging findings. Imaging using mainly echograms and tomodensitometric scans were regularly performed: echograms every 6 months, and tomodensitometric scans every 1 to 2 years. Five groups of patients were identified: those with a normal pancreas (n = 4), incomplete lipomatosis of the pancreas (n = 9), complete lipomatosis of the pancreas (n = 23), cystic pancreas (n = 5 as in figure 5), macrocystic pancreas (n = 1), atrophic pancreas (n = 13). Forty episodes of pancreatitis were observed in seven patients – they had bouts of abdominal pain and elevation of lipase levels.
Jean Feigelson (Fig. 4a) suggests that studies of pancreatic function should be performed routinely in cystic fibrosis, especially in those with pancreatic sufficiency or in patients with normal pancreatic images. Acute pancreatitis was common and should be diagnosed and properly identified to be differentiated from other acute abdominal syndromes occurring in cystic fibrosis a very important point.
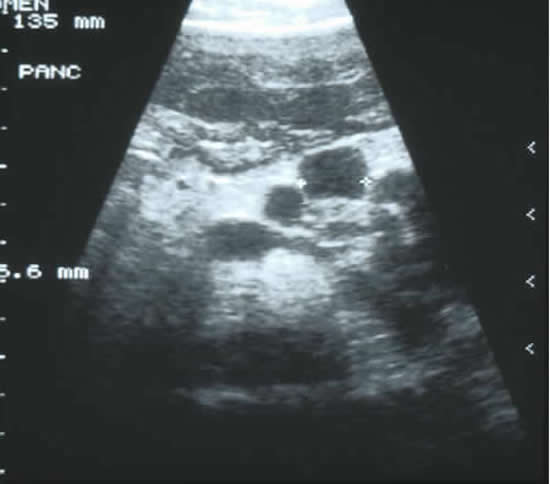
Fig. 5: Abdominal ultrasound showing pancreatic cysts in an adolescent boy with cystic fibrosis
However, it is unlikely that the pancreas will receive such regular and careful attention in many CF centres where more attention is given to the chest than the pancreas and even simple regular evaluation of intestinal fat malabsorption is the exception rather than the rule.
Fogarty A, Hubbard R, Britton J. International comparison of median age at death from cystic fibrosis. Chest 2000; 117:1656-1660. [PubMed]
Data from ten countries in North America, Europe, and Australasia of all persons registered as having died of CF from 1980 to 1994 were analysed by Prof. John Britton and his colleagues from Nottingham, England. The overall median age at death increased from 8 years in 1974 to 21 years in 1994. Median age at death also increased within all countries and was consistently highest in the United States. Women were significantly more likely to die at a younger age than men.

Fig. 6: John Britton CBE. From health.parlicentre.eu
The median age of death is one of the basic indicators of survival. It is encouraging that the median age of death steadily increases each year as the effect of better treatment is reflected in improved survival. The more recent figures for median age of death for patients registered with the UK CF Patient Registry are encouraging – in 2002 the median age of death was 23 yrs, in 2003 24.2 yrs and in 2004 25.6 years, in 2008 27 years and in 2010 it had risen to 29 years.
Professor John Britton (fig. 6) is Professor of Epidemiology, Division of Epidemiology and Public Health, University of Nottingham. He is based in the City Hospital where the Nottingham Adult and Paediatric CF Units are situated.
Gee L, Abbott J, Conway SP, Etherington C, Webb AK. Development of a disease specific health related quality of life measure for adults and adolescents with cystic fibrosis. Thorax 2000; 55:946-954. [PubMed]

Fig. 7 Janice Abbott
Health related quality of life (QOL) measurement is regarded as important in determining the impact of disease on daily functioning and subsequently informing interventions. The questionnaire described here is a fully validated disease specific measure consisting of 52 items across nine domains of functioning which have been identified by, and are of importance to, adolescents and adults with cystic fibrosis.
This is a useful measure for clinical trials and longitudinal studies and QOL measurements and these are required by many journals where patient outcomes are featured (also Quittner AL et al. Translation and linguistic validation of a disease-specific quality of life measure for cystic fibrosis. J Pediatr Psychol 2000; 25:403-414.[PubMed]).
Professor Jan Abbott (fig. 7) of the Faculty of Health, University of Central Lancashire, Preston UK. Over the past 20 years Jan Abbott has made major contributions to the CF care and understanding of the psychosocial aspects of people with CF. She has been involved in many research studies with colleagues at major CF centres such as Manchester and Leeds. Studies have concerned matters relating to treatment adherence, personal and family functioning, eating behaviour and increasingly with quality of life issues.
Ghose I, Mason GC, Martinez D, Harrison KL, Evans JA, Ferriman EL, Stringer MD. Hyperechogenic fetal bowel: a prospective analysis of sixty consecutive cases. BJOG: An International Journal of Obstetrics & Gynaecology 2000; 107:426-429. [PubMed]
A two year prospective analysis of all second trimester fetuses (16-22 weeks of gestation) with hyperechogenic bowel was undertaken. Hyperechogenic fetal bowel (sonographic pathogenicity similar to or greater than that of surrounding fetal bone) was diagnosed using strict criteria. Outcome of affected fetuses was ascertained from hospital records, health care workers and autopsy reports, up to six months of age.
Sixty consecutive fetuses were identified, of which 48 (80%) were live born. Six pregnancies were terminated, four ended with an intrauterine fetal death and two died at birth. The incidence of cystic fibrosis and aneuploidy were each 5%, and there were no cases of congenital infection. Intrauterine growth retardation was recorded in six fetuses (10%), four of whom died perinatally.
So eighty-two percent of fetuses (28/34) with isolated hyperechogenic bowel had a normal outcome. A useful record of practical experience when considering this not infrequent question of the significance of an hyperechogenic fetal bowel.
Gilljam M, Antoniou M, Shin J, Dupuis A, Corey M, Tullis DE. Pregnancy in cystic fibrosis. Fetal and maternal outcome. Chest 2000; 118:85-91.[PubMed]
From 1963 to 1998, there were 92 pregnancies in 54 women known to the Toronto clinic. There were 11 miscarriages and 7 therapeutic abortions. Forty-nine women gave birth to 74 children. The overall mortality rate was 19% (9 of 48 patients). Absence of Burkholderia cepacia (p < 0.001), pancreatic sufficiency (p = 0.01), and pre-pregnancy FEV1 > 50% predicted (p = 0.03) were associated with better survival rates. When adjusted for the same parameters, pregnancy did not affect survival compared to the entire adult female CF population.
The maternal and fetal outcome is good for most women with CF in this major CF centre for adults in Toronto. Good communication between the CF team and the obstetrician is important. Both these papers emphasise the need to plan pregnancies, that severity of the mothers’ chest determines the maternal outcome but that most infants are normal. The women should be closely supervised by the team at a specialist CF centre working closely with an obstetrician with experience in CF pregnancies. Ideally the woman with CF should discuss with the CF team, particularly the dietitian, before becoming pregnant.
Dr Elizabeth Tullis (fig. 8) is Head, Division of Respirology and Director Adult Cystic Fibrosis Program, St Michael’s Hospital, Toronto. She is Director of the largest Adult CF centre in North America providing care for 350 adults with cystic fibrosis.

Fig. 8:Elizabeth Tullis
Goldstein AB, Goldstein LS, Perl MK, Haug MT, Arroliga AC, Stillwell PC. Cystic fibrosis patients with and without central nervous system complications following lung transplantation. Pediatr Pulmonol 2000; 30:203-206. [PubMed]
Eleven of 21 patients (52%) with CF had CNS events after lung transplantation: eight had seizures, five severe headaches, three had strokes, and one had a confusional episode. The authors could not identify any predictive risk factors but considered cyclosporine toxicity to be the major cause of the CNS complications. Despite the high rate of CNS events, the overall prognosis for these patients was good, and their 6-month survival was not affected.
– A surprisingly high percentage of people with CF have neurological problems after lung transplantation. CNS complications are not common in CF but Arnold-Chiari malformations have been reported (Needleman JP et al. Pediatr Pulmonol 2000; 30:490. above [PubMed]).
Hyde SC, Southern KW, Gileadi U, Fitzjohn EM, Mofford KA, Waddell BE, Gooi HC, Goddard CA, Hannavy K, Smyth SE, Egan JJ, Sorgi FL, Huang L, Cuthbert AW, Evans MJ, Colledge WH, Higgins CF, Webb AK, Gill DR. Repeat administration of DNA/liposomes to the nasal epithelium of patients with cystic fibrosis. Gene Ther 2000; 7:1156-65. [PubMed]

Fig. 8a Kevin Southern
A double-blinded study in which two doses of a DNA/liposome formulation were delivered to the nasal epithelium of 10 people with CF; two received placebo. On average, six of the treated subjects showed evidence of CFTR gene transfer after each dose. All subjects who were positive for CFTR function was also positive for plasmid DNA, plasmid-derived mRNA and CFTR protein in the nasal epithelial cells.
The efficacy measurements suggest that, unlike high doses of recombinant adenoviral vectors, DNA/liposomes can be successfully re-administered without apparent loss of efficacy; this was the first study to show this. However, as in all previous studies, the correction was not of a degree likely to influence the clinical state.
Steve Hyde and Deborah Gill became the leaders of the Oxford group of the UK Gene Therapy Consortium. The clinical aspects of the study were coordinated by Dr Kevin Southern (Fig. 8a) as part of his PhD thesis. Kevin later became Reader then Professor in Paediatrics in the Liverpool University Department of Paediatrics at the Alder Hey Children’s Hospital.
Howard M, Frizzell RA, Bedwell DM. Aminoglycoside antibiotics restore CFTR function by overcoming premature stop mutations. Nature Med 1996; 2:467-469.

Fig 9. David Bidwell
Dr David Bedwell (fig. 9) is Professor of Microbiology University of Alabama and Birmingham. His team are exploring whether pharmacological agents can be used to suppress nonsense mutations that cause genetic diseases – “CF is caused by mutations in the CFTR gene (which corresponds to the mouse Cftr gene). We have published several papers demonstrating that drugs can suppress nonsense mutations in the CFTR gene in various CF experimental systems, including cultured CF cell lines and a CF mouse expressing a human CFTR-G542X transgene. Most recently, we constructed a new Cftr-G542X knock-in mouse model to explore this approach in a more physiologically relevant context”.
Subsequently, Bedwell and co-workers showed in a CF bronchial epithelial cell line carrying the CFTR W1282X premature stop mutation, that gentamicin was capable of restoring CFTR expression on the apical membrane. Bedwell DM, Keanjak A, Bebok Z, Bubien JK, Tousson A, Clancy JP, Sorscher EJ. Suppression of a CFTR premature stop mutation in a bronchial epithelial cell line. Nature Med 1997; 3:1280-1284. [PubMed].
Intravenous gentamicin was also capable of producing small increases in CFTR conductance as judged by nasal PD measurements.
Clancy JP, Bebok Z, Ruiz F, King C Jones J, Walker L, Hong J, Wing L, Macaluso M, Lyrene R, Sorscher EJ, Bedwell DM. Evidence that systemic gentamicin suppresses premature stop mutations in patients with cystic fibrosis. Am J Resp Crit Care 2001; 163: 1683-1692.[PubMed].
Further work from Israel on this subject.
Hadfield PJ, Rowe-Jones JM, Mackay IS. The prevalence of nasal polyps in adults with cystic fibrosis. Clin Otolaryngol Allied Sci 2000; 25:19-22. [PubMed]

Fig. 9a. Pandora Hadfield Nuffieldhealth
The prevalence of nasal polyps in this endoscopic study of 211 adults with CF was 37%. The commonest nasal symptoms were discharge, headache and obstruction. Intranasal endoscopy usually demonstrated mucosal oedema and thin, clear discharge. The prevalence of allergy, as diagnosed by skin prick testing, was almost twice that of the general population. Tympanometry showed that middle ear effusion was uncommon in these patients.
Surprisingly, previous studies have also shown a low incidence of middle ear infection in people with CF (also Hadfield et al. 2000. below). Endoscopic studies are required to give an accurate estimate of the incidence of nasal polyps that may have been under-estimated in previous studies.
Pandora J Hadfield (fig. 9a) in an ENT surgeon specialising in children’s and adult’s ENT problems.
Hadfield PJ, Rowe-Jones JM, Mackay IS. A prospective treatment trial of nasal polyps in adults with cystic fibrosis. Rhinology 2000; 38:63-65. [PubMed]
The first prospective, randomised, double-blind trial of the treatment of nasal polyps in cystic fibrosis. Use of betamethasone nasal drops showed a statistically significant reduction in polyp size in comparison to placebo treatment. This is the first trial of topical steroid treatment of nasal polyps, a complication that can prove a troublesome and recurring problem for the patient. Inhaled steroids appeared to have some effect but they had never been the subject of a clinical trial. The recurrence rate after surgery is very high.
In a more recent study of the non-steroidal anti-inflammatory ibuprofen for CF lung disease, not concerned with nasal polyps, the drug was noted to have a favourable and quite unexpected effect on the nasal polyps. This was noted as an incidental finding (Lindstrom D R, et al. J Otolaryngol 2007; 36:309-314 [PubMed] below). As yet (2023) the favourable effect of ibuprofen had not been confirmed.
Israel NR, Khanna B, Cutler A, Perry M, Caplan D, Weatherly M, Gold BD. Seroprevalence of Helicobacter pylori infection in cystic fibrosis and its cross-reactivity with anti-Pseudomonas antibodies. J Pediatr Gastroenterol Nutr 2000; 30:426-431. [PubMed]
The seroprevalence of H. pylori in a cohort of patients with CF and its cross-reactivity with Pseudomonas antibodies were investigated using competitive inhibition assay. The research ELISA and 3 commercial tests initially showed H. pylori seropositivity of 47%, 28%, 24%, and 37%, respectively but post adsorption seropositivity declined to 8%, 0%, 0%, and 15%, respectively and all these positives were confirmed endoscopically to have H. pylori infection.
So cross-reactivity between solid-phase H. pylori antigens and anti-Pseudomonas antibodies occurs in patients with CF. The authors advise that preadsorption of CF sera with Pseudomonas proteins should be used in serologic testing for H. pylori.
– This was a useful study as there had been confusion about the prevalence of H. pylori in people with CF since the existence of cross reactivity with Pseudomonas antibodies had been reported from Niels Hoiby’s laboratory in Denmark (Johansen HK et al. Clin Diag Lab Immunol 1995; 2:149-155.[PubMed]).
One might perhaps have predicted the expected the prevalence of H pylori in people with CF to be low (8-15%) in view of the numerous courses of antibiotics they receive! This and subsequent studies (Yahav J et al. Dig Dis Sci 2006; 51:2274-2279. [PubMed]) confirm that H. pylori is not a significant problem for people with CF.
Jakobsson B, Hjelte L, Nyström B. Low level of bacterial contamination of mist tents used in home treatment of cystic fibrosis patients. J Hosp Infect 2000; 44:37-41. [PubMed]
Mist tents are recommended by the Stockholm cystic fibrosis centre for small children with CF. The disinfection procedures recommended are reviewed – including by home visits. No or insignificant growth was found in 16/20 aerosol tubes: moulds in three, Pseudomonas species in one. Twelve of 19 chambers showed no or insignificant growth: moulds or yeasts were present in three and Pseudomonas sp. in four. In four of the seven patients moulds or yeasts and/or Pseudomonas sp. grew both from chambers and from aerosol tubes; in the remaining three only from chambers. None of these seven patients had followed the prescribed cleaning and disinfection recommendations.
The authors conclude that their disinfection recommendations are adequate when followed. The different forms of non-compliance would not have been detected without a home visit, emphasizing the importance of such visits. The importance of drying the equipment and of using the correct concentration of acetic acid is stressed.
– It is interesting that mist tents are still used in Stockholm although abandoned during the Seventies in most CF centres. It was always a concern that they may be a source of infection if not meticulously cleaned. Before dismissing the use of mist tents it is worth considering the increasing support for the importance of the fluid layer of the respiratory epithelium and also Robert Wood’s comments that he had observed a considerable amount of moisture in the airways when bronchoscoping patients soon after mist tent therapy.
(Suggest see the Topic section on Mist Tents where there is an excellent review by M L Labonte)
Kariyawasam HH, Pepper JR, Hodson ME, Geddes DM. Experience of totally implantable venous access devices (TIVADs) in adults with cystic fibrosis over a 13-year period. Respir Med 2000; 94:1161-1165. [PubMed]
A report of extensive experience of totally implantable venous access devices (TIVADs) in patients with CF over a 13-year period. The patients were divided into those who had the device inserted at the Royal Brompton Hospital (RBH), London and those who had the device inserted elsewhere; however all the devices were cared for at the RBH. A total of 115 devices in 74 patients were reviewed. The median duration of function of 109 devices was nearly 4 years (1429 days – range 2-3989) with a total exposure of 91,188 days or 249.8 years. Forty patients had complications in 62 devices.
The incidence of complications was 34.5% at the devices inserted at RBH and 73.7% for those inserted elsewhere (P<0.001). Of the 115 devices, mechanical complications occurred in 42 (36%), infectious complications occurred in 16 (14%) and symptomatic venous thrombosis occurred in four (3.5%). One of the early reports on TIVADs in people with CF came from the adult CF unit at the Brompton Hospital in 1987 (Stead RJ et al. Thorax 1987; 42:149-150. [PubMed]).
The authors of the present paper conclude that TIVADs provide effective and long-term intravenous access and have fewer complications if they are inserted and cared for at a CF centre with special expertise in their insertion and management. This is the general reported experience at other large centres (Aitken ML. Tonelli MR. Chest 2000; 118:1598-1602. [PubMed]) and also our experience in Leeds.
Keevil B, Rowlands D, Burton I, Webb AK. Assessment of iron status in cystic fibrosis patients. Ann Clin Biochem 2000; 37:662-665. [PubMed]

Fig. 9b Brian Keevil researchgate
The use of soluble transferrin receptor (sTfR), together with more traditional measurements such as iron, transferrin and ferritin, were used to assess iron status in 70 adult CF patients. Sixty nine percent of subjects as determined by transferrin saturation had iron deficiency, but only 29% as determined by sTfR and only 11% as determined by ferritin. There was a significant correlation between C-reactive protein (CRP) and both ferritin and transferrin saturation.
Because the CRP concentration was elevated in 64% of subjects, it may be that the transferrin saturation was overestimating, and the ferritin underestimating, iron deficiency in these patients. The sTfR concentration, on the other hand, is unaffected by the acute-phase response and was therefore thought to be the most useful test for detecting iron deficiency in this group of patients.
Prof. Brian Keevil (Fig. 9b) is at the Department of Clinical Biochemistry, South Manchester University Hospital (NHS) Trust, UK.
– There is an extensive literature on iron in cystic fibrosis dating back to the early Sixties since Davis and Badenoch reported increased iron absorption in pancreatitis (Lancet 1962; 2:6), a finding confirmed in children with CF (Smith RS. BMJ 1964; 1:608-609. [PubMed]). Subsequently a number of studies found low iron status in people with CF including those in Leeds where 41 (32%) of 165 patients had low ferritin levels (Ehrhardt P et al. Arch Dis Child 1987; 62:185-187. [PubMed]). More recently Fischer R et al (Pediatr Pulmonol 2007; 42:1193-1197[PubMed] ) found no benefit from iron supplementation and concluded that chronic inflammation was the main cause of the low iron status. This seems to be the most likely explanation although it seems the matter is still unresolved. There has been discussion regarding bacterial growth and iron. Subsequent publications have failed to provide additional information. Uijterschout L et al conclude their study “shows that ID is common in relatively healthy, well-nourished children with CF. The mechanism of ID in children with CF is currently unknown. A prospective study using both soluble transferrin receptor and Fer as indicators for ID will provide more insight in the incidence and causes of ID in children with CF” (Pediatr Pulmonol 2014:49(5):458-62) pubmed.ncbi.nlm.nih.gov/24000193/)
Krebs NF, Westcott JE, Arnold TD, Kluger BM, Accurso FJ, Miller LV, Hambidge KM. Abnormalities in zinc homeostasis in young infants with cystic fibrosis. Pediatr Res 2000; 48:256-261. [PubMed]

Fig. 9c Nancy F Krebs Googlescholar
Low plasma zinc concentrations have been reported in approximately 30% of young infants with CF identified by newborn screening. This study examined zinc homeostasis in this population by application of stable isotope methodology. Faecal zinc losses correlated with faecal fat excretion, suggesting interference with the normal conservation of endogenously secreted zinc suggesting impaired zinc homeostasis and suggest an explanation for the reported suboptimal zinc status in many young infants with CF prior to diagnosis and treatment.
Prof. Nancy F Krebs (Fig. 9c) is Head, Section of Nutrition, Department of Pediatrics, University of Colorado School of Medicine, Denver 80262, USA.
– The results of many studies on zinc status are conflicting. In 1986 the late Jerry Kelleher reported the zinc status of 117 children with CF in Leeds and found their levels, except for two children, to be quite normal (Kelleher J et al. Hum Nutr Appl Nutr 1986;40A:79-84. [PubMed]). In a later study Ansari et al.1999 (above), as part of a study of vitamin A, also found the zinc levels were normal in Leeds patients. Studies as recent as 2021 (Bauer SE et al) reported “Low sZn occurs in one-third of children with CF in the first 3 years of life. Cross-sectional and longitudinal analyses revealed discrepant associations between sZn and growth. Therefore, prospective studies are needed to understand the role of Zn in growth in CF.” pubmed.ncbi.nlm.nih.gov/34499426/
Kostuch M, Semczuk A, Szarewicz-Adamczyk W, Gasowska-Giszczak U, Wojcierowski J, Kulczycki L. Detection of CFTR gene mutations in patients suffering from chronic bronchitis. Arch Med Res 2000; 31: 97-100. [PubMed]
The purpose of this study from Poland was to search for CF gene mutations in patients suffering from chronic bronchitis. No less than five of 32 (16%) patients admitted to the Lublin School of Medicine, Poland between 1995 and 1996 with chronic bronchitis had CF gene mutations, all within the DeltaF508 region of the CFTR gene. All positive samples were obtained from patients heterozygous for the DeltaF508 mutation. The presence of the DeltaF508 mutation was considered statistically significant when the study group was compared to the study of Poland’s general population. The results suggested an increased presence of the DeltaF508 mutation in Polish patients suffering from chronic bronchitis although the numbers in the study are very small.
M Kostuch is in the Department of Human Genetics, Lublin School of Medicine, Lublin, Poland.
– Earlier clinical studies from the UK, admittedly only using Shwachman finger print plates, did not show people with unrecognised or mild CF were present among patients considered to have chronic bronchitis (Muir D et al. 1962 above). Also in another study, perhaps more relevant to the present report, there was no increase in the frequency of chronic bronchitis among obligate CF heterozygotes (Batten et al, 1963 above). However, the prevalence of chronic bronchitis was very high in the UK general population at that time (presumably due to the high proportion of smokers and atmospheric pollution in London) and this may have obscured the influence of the CF carrier state.
More recently complete mutational screening of 120 patients with chronic pulmonary disease detected mutations in 11 of 23 with disseminated bronchiectasis, 7/25 emphysema, 5/27 chronic bronchitis, 5/26 lung cancer, 5/8 sarcoidosis (Bombieri et al, 1998 [PubMed]). In a more recent study 17.74% of 31 patients with chronic obstructive pulmonary disease had a CFTR mutation (Divac et al, 2004. [PubMed]). Although others searching for only the six common mutations found no increase in CFTR mutations in 100 patients with chronic bronchitis (Entzian P et al, 1995. [PubMed]). However, 5/11 non-CF individuals who had allergic bronchopulmonary aspergillosis (ABPA) had one CFTR mutation (Miller PW et al, 1996. [PubMed]). Also CFTR function has been reported as significantly depressed in smokers (Cantin AM et al. Am J Resp Crit Care Med 2006; 173:1139-1144. [PubMed]).
Lai HC, FitzSimmons SC, Allen DB, Kosorok MR, Rosenstein BJ, Campbell PW, Farrell PM. Risk of persistent growth impairment after alternate-day prednisone treatment in children with cystic fibrosis. N Eng J Med 2000; 342:851-859. [PubMed]

Fig. 9e Hui-Chan Lai nutrisci.wisc.edu
The growth 6 to 7 years after prolonged alternate-day treatment with prednisone had been discontinued, was recorded in 224 children aged 6 to 14 years with CF who had participated in a multicenter trial of this therapy from 1986 to 1991 (a treatment first reported in the paper of Auerbach et al, 1985 above). The Z scores for height of the treated patients declined during prednisone therapy; although catch-up occurred, the mean heights for boys 18 years of age or older were 4 cm less in the prednisone groups than in the placebo group, an equivalent of 13 percentile points. Among the girls, differences in height between those who were treated with prednisone and those who received placebo were no longer present two to three years after prednisone therapy was discontinued.
Hui-Chan Lai (Fig. 9e) is Professor of Nutritional Sciences, Pediatrics, and Population Health Sciences, University of Wisconsin.
– So amongst children with CF who received alternate-day treatment with prednisone, boys, but not girls, had persistent growth impairment after the treatment was discontinued. This was the final chapter in the long term alternate day prednisone treatment in CF first reported in CF by Auerbach et al in 1985 (above). At the time, there had been high hopes that the prednisone treatment would favourably modify the course of the chest infection by significantly suppressing the excessive damaging inflammation in the airways. Prednisolone was used in shorter studies by Pantin et al who showed no significant effect (Thorax 1986; 41:34-38. above [PubMed]) and Greally P et al. who showed over 12 weeks there was improved respiratory function and some reduction in inflammatory markers (Arch Dis Child 1994; 71:35-39. [PubMed]).
Littlewood JM, Koch C, Lambert PA, Hoiby N, Elborn JS, Conway SP, Dinwiddie R, Duncan-Skingle F. A ten year review of colomycin. Respir Med 2000; 94:632-640. [PubMed]
A review of colomycin use in CF up to that time. In the concluding section it was noted that colomycin had re-emerged as the first choice antibiotic treatment for nebulised treatment of both early (Littlewood et al. 1985 above; Valerius et al. 1991 above) and chronic P. aeruginosa infection (Jensen et al, 1989 above) in people with CF certainly in Europe if not N. America where tobramycin was preferred. Colomycin was also a valuable addition to the range of IV antibiotics used for P. aeruginosa when the organism becomes resistant to the more frequently use drugs tobramycin and ceftazidime. It seemed likely to the authors of this review that the antibiotic would continue to have an important role in the treatment of people with CF during the next decade – subsequently this proved to be the case.
Littlewood JM. Good care for people with cystic fibrosis. Paediatr Resp Rev 2000; 1(2):135-140. [PubMed]

Fig. 10 Jim Littlewood
The improvement in the health and survival of people who have cystic fibrosis has, up to the present, been due to better treatment developed at major CF centres. The regimens for prevention, early treatment and later stabilization of chronic respiratory infection and for the maintenance of normal nutrition and growth are now largely established. Treatment is life-long, complex and expensive. It should be started early after a diagnosis made following neonatal screening, and before chronic respiratory infection and malnutrition are established. Regular monitoring and input from the expert staff of a CF centre is essential, either on a ‘full’ or ‘shared care’ basis; adults with CF should attend a major Adult CF unit.
The details of the staff and facilities necessary to achieve good care for CF are discussed in this article, including the details of clinic procedures and annual assessments and based on experience treating some 600 patients at the Leeds Regional CF Centre. Based on this experience I unashamedly detail in the following tables the factors I consider of importance in the provision of good care to people with cystic fibrosis or indeed many other serious chronic disorders!!
These tables below are expanded from those in this article. Also I asked a number of respected experienced UK colleagues to review the tables and received a number of helpful suggestions. The concept of “fatal flaws” of the CF Foundation were gained from a visit to a meeting of the CFF Centres Committee, to which I was invited by Bob Beall. A “fatal flaw” required correction before further funding would be allocated to the centre from the CF Foundation.
Figure 10: Some features of best care for people with cystic fibrosis some of which may not appear in Standards and Guidelines documents
(CFF) = CF Foundation’s ‘Fatal Flaws’
BEST CARE |
SUBOPTIMAL CARE |
Diagnosis |
|
| – Antenatal screening & Diagnosis | No antenatal screening or diagnosis |
| – Routine neonatal screening | No neonatal screening |
| -Single person receives positive screening result and deals with family and coordinates the subsequent investigation. One person responsible. Ideally at a CF centre | Confusion and multiple people, such as the family doctor and inexperienced junior doctors involved who have little recent knowledge of CF |
| – Early diagnosis – before symptoms. The weight falls off within weeks | Late, after prolonged symptoms, chronic lung damage and malnutrition |
| – Rapid, confident & correct diagnosis and early treatment and advice by experts | Slow, unsure, sometimes incorrect by inexperienced people |
| – Good, effective communication by staff experienced in CF | Poor, ineffective communication by staff inexperienced in CF care |
| – Reliable sweat tests even on infants | Unreliable sweat tests
(CFF) |
| – Genotype always determined | Genotype unknown |
| – Siblings all checked for CF | Siblings not checked for CF |
| – Genetic advice for parents & relatives particularly those of child bearing age | No genetic advice to family or relatives |
CF Centre / Clinic |
|
| – Segregation of all patients according to their microbiological status | No segregation of patients according to microbiology |
| – High standards of general hygiene | Poor hygienic standards – common in UK |
| – More than 50 patients on full care | Fewer than 50 patients on full care
(CFF) |
| – Adequate clinic session time identified for CF patients only | Seen in an overcrowded general clinic
(CFF) |
| – Regular follow-up (every 1 to 2 months) | Infrequent, often missed, follow-up |
| – A few permanent CF doctors familiar with each patient’s details | Changing doctors, unfamiliar with CF and the patients they are seeing |
| – One named senior CF doctor (consultant) ultimately responsible for long term care | Unclear who in the “team” is ultimately responsible |
| – Experienced permanent clinic nurses who know patients, cross infection problems, weighing and measuring, spirometry and taking throat swabs and various treatment protocols | Inexperienced temporary nurses who are unfamiliar with the clinic routines |
| – CF Nurse Specialist available if required | No CF Nurse Specialist available or one who is heavily overworked |
| – Usually sees CF Physiotherapist or can have an early appointment as needed | Rarely or never sees Physiotherapist |
| – Usually sees CF Dietitian or early appointment as needed | Rarely or never sees Dietitian |
| – Clear detailed clinic records & data sheets | Brief illegible medical notes |
| – Accurate weight and height, charted at every attendance | Inaccurate, irregular or not charted weights and heights |
| – Computer print out of or access to previous important basic respiratory and nutritional data readily visible in notes – print out or computer | Doctor searches through disorganized notes for information of previous data every time patient attends |
| – Accurate, bacteriologically clean, spirometer used at every attendance | Infrequent, inaccurate, unclean or no spirometry |
| – Sputum/throat swab cultures every attendance and also when unwell. – Arrangements to send to lab’ by post | Infrequent sputum/throat swab cultures |
| – Patient asked to ring if doesn’t hear culture result – rapid treatment of positive cultures | No action on positive culture for weeks until next clinic |
| – Patients/parents should be aware of their microbiology and its significance | Patients/parents unaware of their microbiology |
| – Annual chest X-ray & whenever needed | Chest X-rays rarely done – “chest clear – doesn’t need an X-ray” |
| – Antibiotic treatment at first sign of even a viral respiratory tract infection (RTI). | Treatment only when RTI well established and obviously progressing – has to prise antibiotic out of doctor. |
| – Patient/parents have prescription for an antibiotic or an antibiotic course at home to use when required | Difficulty getting extra antibiotic with colds etc. |
| – Vigorous eradication policy for early Pseudomonas aeruginosa positive cultures. Known to all staff. | No clear policy of early Pseudomonas eradication |
| – Early resort to IV antibiotics with option of home IV antibiotics | IV antibiotics used only as a last resort |
| – Annual Review completed each year | No or incomplete Annual Review |
| – Centre Director personally discusses Annual Review results with parents/patient | Junior doctor discusses results with parents/patient |
| – Centre Director or CF Consultant writes Annual review letter to family doctor with recommendations | Junior doctor rather than Director does letter with recommendations |
| – Copy of letter goes to patient/parents | Patient/parents not informed of results |
| 24-hour access to a member of the CF team for advice | No 24-hr direct access to CF team
(CFF) |
| – Regular review of eligibility for TOBI, Pulmozyme and ventilatory support | Patients missing out on effective treatments which may help them |
| – Direct access to Director / Consultant with phone number | Senior staff unavailable out of hours |
| -Agreed but flexible transition arrangements to adult CF unit known to parents and patients | Unclear transition policy or no adult CF unit with care continuing n the paediatric unit
(CFF) |
CF Ward Routines |
|
| – Segregation according to microbiological status as in clinic | No segregation according to microbiological status |
| – Highest standards of hygiene for all including all ancillary staff and visitors, cleaners, etc | Poor hygiene standards, inadequate routines & hand washing faculties |
| – Sufficient, knowledgeable ward nurses | Temporary overworked nursing staff |
| – Seen regularly by senior CF doctors | Inexperienced CF naive doctors |
| – Protocols for all routine procedures | No protocols – variable procedures |
| – Single rooms with en suite facilities | Open ward with shared facilities |
| – Good ward cleaning service | Inadequate ward cleaning |
| – Daily visits from CF physiotherapist | No regular physiotherapy |
| – Supervision by CF dietitian | No regular supervision by dietitian |
| – Appropriate, palatable food | Unattractive unpalatable hospital food |
| – Regular ward rounds and information to patient/parents | Irregular ward rounds / inadequate information regarding progress |
| – Education / occupation / interests while in the ward | No education occupation or interests |
| – Communication with other patients via internal phone, internet, etc | Isolation and boredom in cubicle |
| Prevention, Immunization | |
| – Fully immunized against common infectious disease | Incompletely immunized |
| – Annual influenza injections | No influenza injections |
| – Avoidance of people with viral ‘colds’ when possible | Unnecessary exposure to ‘colds’ |
| – Avoidance of tobacco smoke | Smoking in the environment |
| – Avoidance of fungal spores | Indoor work at horse stables |
| – Adequate dry housing | Overcrowded damp house |
| – Avoids others with CF e.g. camps/holidays and social gatherings etc | Attends CF camps/holidays etc |
| – Always avoids others with CF | Mixes freely with others with CF |
| – Adequate personal/family finances | Financial difficulties |
General |
|
| – Patients should be aware of their respiratory function (FEV1 or even PEFR for young patients) and have some idea of where they lie in terms of severity | May be quite unaware of how they are compared to others with CF |
| – All patients severe enough to be in the “transplant window” are reviewed every 6 months | Some patients who reach need for transplant referral are overlooked as need not reviewed regularly |
| – Advice on and aware of state benefits to which they are entitled. Reviewed by the CF social worker | Not receiving full entitlements as not had adequate advice |
| – Aware of and contact with the national CF patients’/parents’ organization (CF Trust) | Unaware of and unknown to national CF organization. |
| – Aware of reliable and useful CF websites (www.cftrust.org.uk;www.cfmedcine.com; www.cff.org) | Unaware of or no access to reliable CF websites |
| – Well informed and “adjusting” to having CF | Ill-informed and not adjusting to having CF |
| – Regular contact with Specialist CF center staff (if a child on ‘shared care’ who is attending the local hospital) | All care at local clinic; irregular or no contact with staff at Specialist CF centre |
| – Registered with UK national CF Database or their national database and regular data entry to local and national database | Not registered with national database and no local database |
Other |
|
| – Close collaboration with Microbiologist and laboratory staff experienced in culturing CF sputum specimens | Routine laboratory with only occasional CF specimens and little contact with clinicians |
| – Efficient centre secretary/coordinator who knows staff and families/patients | Temporary secretarial help to only deal with letters, telephone, appointments, etc |
| – Other laboratories and X-ray departments in the hospital regularly perform specialist CF investigations and procedures | Other departments rarely do investigations on CF patients and so unfamiliar with techniques and likely findings |
| – Help of other colleagues familiar with CF –surgeons, ENT, diabetologist, gastroenterologist, radiologists obstetrician, fertility expert, geneticist, medical physics, pharmacist | Referral to specialists who rarely see people with CF and unfamiliar with particular problems |
| – Suitable funding arrangements agreed with referring health authorities | Constant difficulties prescribing CF drugs and care |
| – Attention to user requirements/ satisfaction/ suggestions | No regular discussion with patients / parents on their views on the adequacy of the CF service |
| – Appropriate management of terminal care and ongoing family support – letters from senior nurse and Centre Director. Possible bereavement counselling | No contact with relatives after patient dies |
| – Attention to the physical and mental well being of all members of the CF team | Overwork, stress, conflict and poor relationships between members leading to inefficient service and illness. Inadequate leadership. |
| – Mandatory weekly meetings of all members of the CF team + microbiologist and pharmacist (when possible) | No regular team meetings to discuss patients, policies and problems
(CFF) |
| – Clear, informed, decisive leadership from the Centre Director – coordinating the individual views into agreed decisions and communal policies. A basis for written protocols. | Independent policies and practises by different team members leading to conflict and confusion for other team members and patients |
| – Consultants and CF team undertake clinical research and present the results at CF meetings and in publications | No research, publications or attendance at CF meetings |
| – Efficient CF Clinic and Data Clerk to ‘guard’ the notes and organise clinics | Reliance on central appointments system & secretarial pool |
| Clinical notes and recent X-rays held in the Cystic Fibrosis Unit | Notes and X-rays in hospital main records office and frequently unavailable |
Mak GZ, Harberg FJ, Hiatt P, Deaton A, Calhoon R, Brandt ML. T-tube ileostomy for meconium ileus: four decades of experience. J Pediatr Surg 2000; 35:349-352. [PubMed]

Fig. 10a Grace Z Mak University of Chicago
A total of 20 of 23 patients had resolution of their meconium ileus after T-tube irrigation with n-acetylcysteine or pancreatic enzymes. Three patients required additional surgery to relieve persistent bowel obstruction. All patients had the T-tube removed within the first 8 weeks after surgery. Two patients required subsequent repair of an incisional hernia. There were otherwise no complications of this procedure, with an average follow-up of 11.5 years. In patients with uncomplicated meconium ileus unrelieved by contrast enema, the T-tube ileostomy is an effective and safe treatment.
– A report of extensive experience of this technique used successfully at Texas Children’s Hospital since 1959.
Grace Z Max (Fig 10a) is now Professor of Surgery and Associate Professor of Pediatrics in the Department of Surgery, University of Chicago
Marchand V, Baker SS, Stark TJ, Baker RD. Randomized, double-blind, placebo-controlled pilot trial of megestrol acetate in malnourished children with cystic fibrosis. J Pediatr Gastroenterol Nutr 2000; 31:264-269. [PubMed]
Megestrol, an appetite stimulant, caused increased weight gain in 6 of the 12 children with CF who completed the trial. Three withdrew for unrelated reasons and three developed diabetes. So megestrol acetate appeared to improve appetite but the side effects were a problem including adrenal suppression, glucose intolerance and diabetes.
Massie RJ, Olsen M, Glazner J, Robertson CF, Francis I. Newborn screening for cystic fibrosis in Victoria: 10 years’ experience (1989-1998). Med J Aust 2000; 172:584-587. [PubMed]
Of 635,157 babies born in Victoria in the 10 years 1989-1998, 191 were diagnosed with CF. A further 30 cases were detected antenatally, giving an incidence of 1/2874. CF was detected early in 182 babies (95.3% of affected babies in the screened cohort)–136 by screening, 35 because they had meconium ileus, and 11 because they were siblings of older children with CF. Nine cases (4.7%) of CF were missed by screening. Of these nine babies, four did not have an elevated neonatal IRT level, one had a normal IRT level at repeat testing at 4-6 weeks (1989-1990), three did not have a delta F508 mutation (1991-1998), and one had a false negative sweat test result. Nevertheless 6 of the 9 missed babies (67%) were diagnosed within four months of birth.
– These are really excellent figures and the authors justifiably concluded that newborn screening for CF in Victoria had proved effective in detecting most babies with CF in the newborn period. However, they stressed that a sweat test should still be requested by doctors when the clinical features suggested the diagnosis of CF, even if the child had been screened as a neonate.

Fig. 11: John Massie
Massie J, Gaskin K, Van Asperen P, Wilcken B. Sweat testing following newborn screening for cystic fibrosis. Pediatr Pulmonol 2000; 29:452-456. [PubMed]
Thirty-nine of 85 DeltaF508 homozygous and 270 of 274 DeltaF508 heterozygous infants had sweat tests reported to the screening program. There were 6 DeltaF508 heterozygous infants with sweat chloride concentrations of 40-60 mmol/L and subsequently 4 of the 6 had CF confirmed. Infants with sweat chloride levels of 40-60 mmol/l require further investigation and review, but they almost certainly have CF.
The chloride/sodium ratio previously recommended in evaluating marginal sweat test results (Green A, et al. Ann Clin Biochem 1985; 22:171-174. [PubMed]; Henderson MJ, et al. Ann Clin Biochem 1986; 23:109 [PubMed]😉 was not useful in establishing a diagnosis of CF in these infants.
– This excellent paper is helpful and important confirmation that sweat chloride results can be well below the traditional level of 60 meq/l for a positive diagnosis in young infants with CF. In a later study of young non-CF infants the sweat chloride was inversely related to age at testing. For example, chloride values for infants aged between 3-7 days were only 23.3 +/-5.7 mmol/l, 8-14 days 17.6 +/-5.6 mmol/l, 15 – 42 days 14.8 +/- 5.9 mmol/l, and more than 42 days 13.1 +/- 7.4mmol/l (Eng W et al. Pediatr Pulmonol 2005; 40:64-67. [PubMed]).
Professor John Massie (fig. 11) trained in Sydney (from where this paper originated) and is now head of Education and Training at the Royal Children’s Hospital, Melbourne.
Moran A. Cystic fibrosis-related diabetes: an approach to diagnosis and management. Pediatr Diabetes 2000; 1:41-48. [PubMed]
The Cystic Fibrosis Foundation held a consensus conference in 1998 to define the current standards for the diagnosis and care of CF related diabetes (Moran A, Hardin D, Rodman D et al. Diagnosis, screening, and management of CFRD: a consensus conference report. J Diabetes Res Clin Pract 1999: 45: 61-73). This article reviews those recommendations, and presents the practical approach to the management of CFRD used at the University of Minnesota.
The CF Foundation recommendations were reviewed in 2009 (Moran A. Updates on CFRD guidelines. Pediatr Pulmonol 2009; Suppl 32:208-209. S19.4). There was a further CFF Consensus Conference on CFRD in Sept. 2009.

Fig 12: Antoinette Moran www.med.umn.edu
Dr Antoinette Moran (figure 12) of the University of Minnesota was awarded the CF Foundation’s Distinguished Clinical Achievement Award in 2007 for her work in this area.
The UK CF Trust consensus document Management of Cystic Fibrosis Related Diabetes Mellitus by the CF Trust’s Diabetes Working Group was published in 2004. Diabetes mellitus is one of the major age-related complications which is likely to affect the majority of adults with CF eventually.
Mekus F, Ballman M, Bronsveld I, Bijman J, Veeze H, Tummler B. Categories of DF508 homozygous cystic fibrosis twin and sibling pairs with distinct phenotypic characteristics. Twin Res 2000; 3:277-293. [PubMed]

Fig. 12a Frauke Mekus ecorn-cf
A large European twin study searching for features for which the twins were discordant. Monozygotic twins with CF had less discordance than dizygotic twins indicating that, in part, CF severity is modulated by an inherited component in addition to the CFTR itself and the very important environmental factors.
Although there was increasing interest in the effect of modifying genes that affected the course of CF in affected individuals, in practical terms the influence of most was relatively small when compared with the influence of the standard of treatment the patient received – or did not receive as the case may be.
Dr Frauke Mekus (Fig. 12a) is in the Department of Pediatrics, Medizinische Hochschule Hannover, Germany.

Fig. 14 Manfred Ballman www.ecfs.en/

Fig.13 Burkhard Tümmler
www.mhh.de
Professor Burkhard Tummler (figure 13) is head of the Clinical Research Group “Molecular pathology of cystic fibrosis”, at the Medical University, Hannover and professor at the Pediatric Clinic of the Medical School of Hannover.
Professor Manfred Ballman (figure 14) is based at the Department of Paediatric and Adolescent Medicine Bochum. He has served on the Board of the European CF Society and is actively involved with national and international CF care and research.
Mitchell I, Nakielna E, Tullis E, Adair C. Cystic fibrosis. End-stage care in Canada. Chest 2000; 118:80-84. [PubMed]

Fig. 14a Ian Mitchell policy options.irpp.org
The mean age at death of the 45 Canadian individuals included in this study was 25.8 years (in UK in 2002 median age of death was 23 yrs, in 2003 24.2 yrs, in 2004 25.6 years and in 2008 27 years). The major cause of death was respiratory failure and lung transplantation was considered in 42 patients (93.2%), with 7 patients (17.1%) being entered on a list, but it was carried out in only 2 patients (4.4%). Autopsies were performed on only 10 patients (22.2%). Most patients died in hospital (37 patients; 82.2%), and 7 patients (15.6%) died in intensive care units while receiving intermittent positive-pressure ventilation. Palliative care was never discussed in 10 patients (25%); in a further 16 patients (40%), it was not discussed until the last month before death. Although lung transplantation is frequently considered and requested, many patients still die without having the opportunity of a transplant. In the UK this is solely due to the shortage of donor organs.
These authors considered that discussions on end-of-life care could be considered sooner. In comparison with the earlier reports there were few autopsies performed. Our own view on this was that there was ample information on the likely findings in the past literature and the procedure would only cause further distress for both the relatives and professional carers – so in Leeds we rarely suggested an autopsy to the families. The need to discuss these end-of-life issues is rightly emphasised as some adults with CF do not realise the serious stage their condition has reached, showing genuine surprise when the subject of transplantation is raised.
Prof. Ian Mitchell (fig.14a) is Professor of Paediatrics University of Calgary.
Mickle JE, Cutting GR. Genotype-phenotype relationships in cystic fibrosis. Med Clin North Am 2000; 84:597-607. [PubMed]
The fact that variability is observed among people with the classic form of CF is evidence that other factors are important. The so-called mild mutations that allow partial function of CFTR are often associated with pancreatic sufficiency, occasionally identified with normal sweat gland function, and sporadically correlated with mild lung disease; also partially functioning mutants rarely prevent maldevelopment of the male reproductive tract; an exception is 3849 + 10 Kb C–>T.

Fig. 15: Garry Cutting. www.hopkinscf.org
These observations suggest that certain tissues require different levels of CFTR function to avoid the pathologic manifestations typical of CF.
However, it is still true that, in the majority of patients, the outlook is more dependent on the standard of treatment the patient receives and whether that treatment is started before there is irreversible lung damage.
From the Department of Pediatrics McKusick-Nathans Institute of Genetic Medicine. Johns Hopkins University School of Medicine, Baltimore.
Professor Garry Cutting (Fig. 15) is interested in the molecular biology and human genetics of ion channel disorders. Much of his work is focused upon cystic fibrosis.
Noone PG, Hohneker KW, Zhou Z, Johnson LG, Foy C, Gipson C, Jones K, Noah TL, Leigh MW, Schwartzbach C, Efthimiou J, Pearlman R, Boucher RC, Knowles MR. Safety and biological efficacy of a lipid-CFTR complex for gene transfer in the nasal epithelium of adult patients with cystic fibrosis. Mol Ther: J Am Soc Gene Ther 2000; 1:105-114. [PubMed]

Fig. 15a Peader G Noone
health.usnews,com
The authors tested the safety and efficacy of a cationic liposome [p-ethyl-dimyristoylphosphadityl choline (EDMPC) cholesterol] complexed with an expression plasmid containing hCFTR cDNA in 11 adults with cystic fibrosis.
Although the lipid-DNA complex was safe, it did not produce consistent evidence of gene transfer to the nasal epithelium either by physiologic or molecular measurements. So this was another gene therapy trial where changes in the epithelial cells were non-existent or inadequate to achieve any clinical benefit.
Dr Paeder G Noone (Fig. 15a) is a pulmonologist in Chapel Hill NC.
Needleman JP, Panitch HB, Bierbrauer KS, Schidlow DV. Chiari type I malformation in children and adolescents with cystic fibrosis. Pediatr Pulmonol 2000; 30:490-492. [PubMed]
Chiari type I malformation is characterized by herniation of the cerebellar tonsils through the foramen magnum (figure 17). Five children and adolescents with CF in whom Chiari type I malformations were diagnosed had neurological problems including swallowing dysfunction, syncopal episodes, numbness of extremities, recurrent vomiting, and headaches.
The authors suggest that Chiari type I malformation is more common in CF than in the general population and advise the possibility should be included in the differential diagnosis of unexplained neurological complaints in people with CF

Fig 16: Daniel Schidlow. www.drexelmed.edu
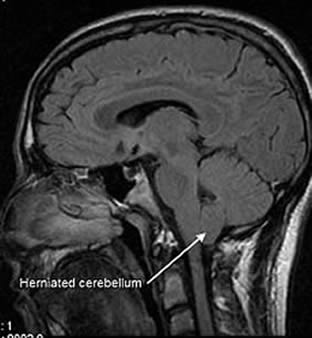
Fig. 17: Arnold-Chiari malformation From Wikipedia
This is a previously unreported and serious neurological complication found in 5 of 400 patients with CF. An important complication of which many paediatricians will have encountered in relation to spina bifida and hydrocephalus but not associated with CF.
A further 2 infants with CF and the Chiari type 1 malformation were reported in 2003 (Rakheja D et al. Pediatr Develop Path 2003; 6:88-93. [PubMed]). The authors commented that the chances of the association occurring by chance was one in 7,500,000 so it was likely that there was some relationship. Also neurological complications are relatively common after lung transplantations (Goldstein et al, 2000. below. [PubMed]).
Dr Daniel Schidlow (fig. 16) was appointed Walter H. and Leonore Annenberg Dean and senior vice president, medical affairs, at Drexel University College of Medicine on June 27, 2012. Dr. Schidlow specializes in cystic fibrosis and pulmonary conditions in children. He has researched and published widely on cystic fibrosis.
Ojeniyi B, Frederiksen B, Hoiby N. Pseudomonas aeruginosa cross-infection among patients with cystic fibrosis during a winter camp. Pediatr Pulmonol 2000; 29:177-181.[PubMed]

Fig.17a Birgitte Frederiksen & Niels Hoiby. author’s photo
In 1990 twenty-seven patients with CF from the Danish Cystic Fibrosis Centre went to a winter camp for a week. The study is based on 22 of these patients. Prior to attending camp, 17 out of the 22 patients harboured Pseudomonas aeruginosa in their sputum, but 5 patients did not. After returning from camp, all 22 patients harboured P. aeruginosa in the sputum. The typing results showed that the 5 CF patients who were free of P. aeruginosa in their sputum prior to the winter camp had acquired P. aeruginosa isolates identical to the P. aeruginosa strains isolated from the other 17 CF patients.
The authors concluded that separate holiday camps based on the infection status of the patients with cystic fibrosis are necessary to avoid cross-infection of patients not infected with P. aeruginosa.
– This is one of the more conclusive reports of people with CF contracting new P. aeruginosa infection during a holiday with others who also have CF and who were already chronically infected. The report was important for at this time there were still experienced and respected CF physicians who questioned the importance of segregation according to microbiological status (Geddes DM. Of isolates and isolation: Pseudomonas aeruginosa in adults with cystic fibrosis. Lancet 2001; 358:522-523.).
Pons G, Marchand MC, d’Athis P, Sauvage E, Foucard C, Chaumet-Riffaud P. The Amiloride-AFLM Collaborative Study Group. Pediatr Pulmonol 2000; 30:25-31. [PubMed]
In this large French study 64 patients with CF, chronically infected with Pseudomonas aeruginosa, received either nebulised amiloride or placebo three times daily for 6 months in addition to their usual treatments. The study failed to demonstrate any significant benefit from the adding the amiloride to their treatment.
This multicentre French study confirmed the lack of significant clinical effect of inhaled amiloride the use of which had been of interest as a treatment particularly since Michael Knowles’s amiloride study in the early Nineties – using amiloride to reduce the excessive sodium absorption from the CF airways (Knowles MR et al. N Eng J Med 1990; 322:1189-1194. above [PubMed]). Apparently the usual preparation of amiloride is rapidly destroyed in vivo. As excessive Na absorption in now considered important in the pathophysiology of the respiratory tract, other ways of inhibiting excessive sodium absorption have replaced amiloride as treatment possibilities.
Longer acting analogues of amiloride may be more effective and subsequently became available such as compound 552-02 (Hirsch AJ et al. J Pharmacol Exp Ther 2008; 325:77-88.[PubMed] below); subsequent development of the compound by Gilead has continued as Gilead GS9411 which entered clinical trials in 2009. In April 2010 Gilead reported that – ” With regards to GS 9411, our epithelial sodium channel blocker or ENaC inhibitor, we have now completed both the single and multiple dose Phase I studies in healthy volunteers. In both studies, we saw increases in serum potassium in patients dosed with GS 9411, which is now supporting advancing this particular compound further”.
– Unfortunately phase III trials that followed did not show significant benefit and development was discontinued.
Rappaz I, Decosterd LA, Bille J, Pilet M, Belaz N, Roulet M. Continuous infusion of ceftazidime with a portable pump is as effective as thrice-a-day bolus in cystic fibrosis children. Eur J Pediatr 2000; 159:919-925. [PubMed]
Continuous intravenous infusion of ceftazidime was just as effective as thrice daily IV administration and the method is preferred by the children. This was to be expected. The method of constant infusion for beta lactam antibiotics had been suggested sporadically over the past 20 years by Prof. Tim David of Manchester and others (Vinks AA et al. J Antimicrob Chemother 1997; 40:125-133.[PubMed]) but never became routine in CF centres. Probably this was, in part, related to the cost of the portable infusion pumps required and also initially their reliability was a problem. Other previous authors also found the method acceptable and as effective as intermittent dosing (Bosso JA, et al. Pharmacotherapy 1999; 19:620-626. [PubMed]).
– Continous infusions is the logical way to administer ceftazidime where steady blood levels should be maintained rather than trying to achieve peak serum levels as is ideal with aminoglycosides. A more recent review of experience with the method in severe pneumonia in non-CF patients concluded that in a small, selected population of adult patients with ventilator associated pneumonia caused by gram-negative bacteria who were treated in a non-randomized, open-label manner, ceftazidime administered by continuous infusion had greater clinical efficacy than ceftazidime administered by intermittent infusion” (Lorente L. Clin Ther 2007; 29:2433-2439.[PubMed]).

Fig. 17b Don Hayes Jr. CincinnatiChildren’s Hospital
In 2008 an important short report using continuous IV ceftazidime and daily IV aminoglycoside eradicated Pseudomonas aeruginosa in a 3 month old CF infant when outpatient eradication treatment had failed (Hayes D Jr et al. Novel approach to the eradication of Pseudomonas aeruginosa in an infant with CF after outpatient treatment failure. Pediatr Pulmonol 2008; 43: 511-513. [PubMed]).
This was a very useful report as in most eradication schemes for Pseudomonas there were still 10-15% where initial eradication therapy failed. To use both these antibiotics in their optimal manner would possibly increase the chances of eradication which, if achieved, would in the long term be well worth all the considerable effort and inconvenience to the patient and family.
Don Hayes Jr. (fig.17b) is a Professor and pediatric and adult pulmonologist in Cincinnati hospitals and the University College of Medicine.
Rodgers HC, Knox AJ, Toplis PJ, Thornton SJ. Successful pregnancy and birth after IVF in a woman with cystic fibrosis. Hum Reproduct 2000; 15:2152-3. [PubMed]
The first woman with CF to be successfully treated with in vitro fertilisation (IVF) after repeated failed attempts at intrauterine insemination. As survival increases, patients with CF are often confronted with reproductive issues. Initial reports gave conflicting advice regarding the outcome of pregnancy in CF; however two large longitudinal studies of pregnancies in CF women suggested that in recent years most pregnancy has little impact on morbidity or mortality (Edenborough et al. 2000 [PubMed]; Gilljam et al. 2000 below). [PubMed]

Fig. 18: Sir Robert Edwards.
From BMJ 2013; 346:f2600
Reduced fertility in women with CF has been described, considered possibly due to thick cervical mucus, and intrauterine insemination has been used to overcome this.
In the present case IVF was used – a technique first described by Professor Sir Robert Edwards (fig. 18) and used successfully in non-CF women 1978.
The present case report was from Respiratory Medicine Unit, City Hospital, Hucknall Road, Nottingham NG5 1PB, UK.
In 2010 Professor Edwards (fig. 18) was awarded the Nobel Prize for his work. The technique, first described by Edwards and Steptoe (Steptoe PC, Edwards RG. Birth after the re implantation of a human embryo. Lancet 1978; 2:366.[PubMed]), has proved valuable for people with CF as representing an essential step in performing pre-implantation genetic diagnosis and also in pregnancies following intracytoplasmic sperm injection of samples from men with cystic fibrosis.
Scotet V, de Braekeleer M, Roussey M, Rault G, Parent P, Dagorne M, Journel H, Lemoigne A, Codet JP, Catheline M, David V, Chaventre A, Dugueperoux I, Verlingue C, Quere I, Mercier B, Audrezet MP, Ferec C. Neonatal screening for cystic fibrosis in Brittany, France: assessment of 10 years’ experience and impact on prenatal diagnosis. Lancet 2000; 356:789-794.[PubMed]

Fig. 18a Virginie Scotet
quest-france.fr
A report of 10 years of neonatal CF screening in Brittany, France, and its impact on prenatal screening of subsequent pregnancies in couples with one affected child. Between 1989 and 1998 118 children with CF were identified, an incidence of 1/ 2913. Thirty nine (34%) of the families identified by neonatal screening opted for subsequent prenatal diagnosis at least once in future pregnancies. Twelve couples would have benefited from this procedure while their first child was still symptom-free. Forty two healthy children were born, and 18 pregnancies were terminated (therapeutic abortion rate of 100%).
The authors showed the feasibility of neonatal screening for CF in Brittany. Through the detection of a large range of mutations, neonatal screening provides the opportunity for more reliable prenatal diagnosis and cascade screening for other family members (Super M et al. 1994. above).
Virginie Scotet (fig 18a) is in charge of this research and based in the Laboratories of Professor Claude Ferec
It was of some concern and somewhat surprising that at the time when this excellent French paper appeared, in 2000, the UK National Screening Committee still did not consider there was sufficient evidence to recommend national CF neonatal screening! Fortunately in 2001, on the decision of the then Health Minister Yvette Cooper, the UK Government agreed to the introduction of neonatal CF screening, eventually implemented throughout the UK by 2007.

Fig. 19: Lisa Saiman. From www.columbia.edu
Saiman L, Macdonald N, Burns JL, Hoiby N, Speert DP, Weber D. Infection control in cystic fibrosis: practical recommendations for the hospital, clinic, and social settings. Am J Infect Contr 2000; 28:381-385. [PubMed]
Very detailed report of the findings of a CFF consensus meeting on infection control which included Europeans Niels Hoiby, Gerd Doering and Jim Littlewood. The great detail in this report is a reflection of the increasing realization that cross infection was a common and potentially harmful occurrence in CF centres.
Dr. Lisa Saiman (fig. 19) is a Professor of Clinical Pediatrics at Columbia University and an Attending Professor of Pediatrics at the Morgan Stanley Children’s Hospital of New York-Presbyterian, Columbia University Medical Center where she has served as the hospital epidemiologist since 1994. She has been the director of the CF Foundation’s CF Referral Center for Susceptibility and Synergy Studies since its inception in 1991. Dr. Saiman served as the co-Principal Investigator of a double-blinded, randomized placebo-controlled, multi-center trial of azithromycin in CF patients infected with Pseudomonas aeruginosa(JAMA 2003; 290:1749-56. below. [PubMed]).
Schneiderman-Walker J, Pollock SL, Corey M, Wilkes DD, Canny GJ, Pedder L, Reisman JJ. A randomized controlled trial of a 3-year home exercise program in cystic fibrosis. J Pediatr 2000; 136:304-310. [PubMed]

Fig. 19a Jane Schneiderman ResearchGate
To evaluate the effects of a 3-year home exercise program on pulmonary function and exercise tolerance in mildly to moderately impaired patients with CF and to assess whether regular aerobic exercise is a realistic treatment option. An improved sense of well-being was reported with exercise and pulmonary function declined more slowly in the exercise group than in the control group, suggesting a benefit for patients with CF participating in regular aerobic exercise (percentage predicted decline in FVC 2.42 vs. 0.25 and FEV1 3.47 vs. 1.46).
– This is one of a number of studies that confirmed exercise was good for people with CF. It was always apparent following patients in the clinic over many years that those people with CF who were very active seemed to remain in better condition which is really not surprising.
Dr Jane Schneiderman (Fig.19a) is an Exercise Physiologist/Registered Kinesiologist in the Div. of Respiratory Medicine at the Hospital for Sick Children in Toronto.

Fig. 19b Rosalind Smyth gosh.nhs.uk
Smyth RL, Croft NM, O’Hea U, Marshall TG, Ferguson A. Intestinal inflammation in cystic fibrosis. Arch Dis Child 2000; 82:394-399. [PubMed]

Fig. 19c Anne Ferguson
There is controversy about whether the inflammatory response observed in the cystic fibrosis (CF) lung occurs secondary to bacterial infection or is caused by a dysregulation of the inflammatory response associated with the basic cellular defect of CF.
To study the inflammatory response the gastrointestinal tract of CF children who had developed fibrosing colonopathy, whole gut lavage was performed on 21 pancreatic insufficient children with CF, who were clinically well, five children with CF who had had fibrosing colonopathy, and 12 controls. Intestinal outputs of plasma derived proteins (albumin, alpha1-antitrypsin, IgG), secretory immunoglobulins (IgA and IgM), cellular constituents (eosinophil cationic protein and neutrophil elastase), and cytokines (interleukin 8 and interleukin1beta) were measured.
Compared to controls, all the 21 CF patients, with no intestinal complications, had increased intestinal outputs of albumin, IgG, IgM, eosinophil cationic protein, neutrophil elastase, interleukin 1beta, and interleukin 8. Similar values were obtained for the CF patients who had had fibrosing colonopathy.
Whole gut lavage is essentially washing out the gut and counting the inflammatory cells in the washings as an indication of the degree of intestinal inflammation. These data suggest that there is immune activation in the gastrointestinal mucosa of all children with cystic fibrosis, which may result from the basic cellular defect perhaps with secondary bacterial infection of the gut. Certainly the children who had previously had fibrosing colonopathy did not appear to have more signs of inflammation than those who had not had the complication.
The finding of evidence of intestinal inflammation is not unexpected as it has been reported, and confirmed, that small bowel bacterial overgrowth affects some 70% of people with CF which is likely to be related to these findings (Bali et al, 1983 above). Also more recent work using capsule endoscopy showed that many patients had diffuse areas of inflammation in the small bowel associated with very high caloprotectin levels confirming the inflammatory response (253+/- 97 ug/g Normal <50) (Werlin S et al. Pediatr Pulmonol 2008; Suppl 31:Poster 610:422; Werlin SL et al. J Pediatr Gastro Nutr 2010; 51:304-308. [PubMed]). Subsequently interest in this area increased progressively throughout the decade.
Turner MA, Goldwater D, David TJ. Oxalate and calcium excretion in cystic fibrosis. Arch Dis Child 2000; 83:244-247.[PubMed]
A patient with CF had repeated calcium oxalate renal stones and prompted the authors to investigate other children for risk factors for this recognised complication of cystic fibrosis. They showed a positive correlation between oxalate excretion and glycolate excretion in children with CF, 21 of 24 of whom had a calcium excretion below the normal range. Hyperoxaluria may reflect the intestinal malabsorption although correlation between excretion of oxalate and glycolate suggests that certainly a portion of the excess oxalate is derived from metabolic processes. The low urinary calcium observed here may protect children with CF from renal stones although these are a relatively common occurrence affecting up to 6.3% of patients (Gibney EM, Goldfarb DS. Am J Kid Dis 2003; 42:1-11. [PubMed]; Terribile M et al, 2006 below [PubMed]).
Wang X. Moylan B. Leopold DA. Kim J. Rubenstein RC. Togias A. Proud D. Zeitlin PL. Cutting GR. Mutation in the gene responsible for cystic fibrosis and predisposition to chronic rhino sinusitis in the general population. JAMA 2000; 284:1814-1819. [PubMed] Eleven of 147 chronic rhino sinusitis (CRS) patients (7.5%) were found to have a CF mutation (DeltaF508, n = 9; G542X, n = 1; and N1303K, n = 1). Diagnostic testing excluded CF in 10 of these patients and led to CF diagnosis in 1. Excluding this patient from the analyses, the proportion of CRS patients who were found to have a CF mutation (7%) was significantly higher than in the control group (n = 2 [2%]; P =.04, both having DeltaF508 mutations). Furthermore, 9 of the 10 CF carriers had the polymorphism M470V, and M470V homozygotes were over represented in the remaining 136 CRS patients (P =.03).
These data indicate that mutations in the gene responsible for CF may be associated with the development of chronic rhinosinusitis in the general population. Another study suggesting that CF heterozygotes or even atypical patients with CF may be over represented in a condition with similar clinical features to CF.
Dr X Wang is at the McKusick-Nathans Institute of Genetic Medicine, CMSC 1004, Baltimore, MD 21287, USA.
Wilschanski M, Famani C, Blau H, Rivlin J, Augarten A, Vital A, Kerem B, Kerem E. A pilot study of the effect of gentamicin on nasal potential difference measurements in cystic fibrosis patients carrying stop mutations. Am J Resp Crit Care 2000; 161:860-865. [PubMed]
This study, by Michael Wilschanski (fig.20) and colleagues from Haddash University Hospital, Israel, was the first to determine if gentamicin in vivo can activate mutant CFTR in CF patients carrying stop mutations as had been suggested by Howard et al, (1996) and Bedwell et al, (1997).
Nine people with CF carrying stop mutations received gentamicin nasal drops for 14 days. The abnormal nasal potential difference improved after the gentamicin treatment suggesting that chloride transport had increased. The authors concluded that gentamicin may influence the underlying chloride transport abnormality in patients with CF carrying stop mutations (i.e. those mutations containing an X).
Aminoglycoside antibiotics can apparently increase the frequency of erroneous insertion of nonsense codons hence permitting the translation of CFTR alleles carrying missense mutations to continue reading to the end of the gene. It is appropriate that this study came from Israel as 64% of people with CF that country have at least one stop mutation.

Fig. 20 Michael Wilschanski. Author’s photo.
SOME OF THE WORK THAT FOLLOWED ON STOP MUTATIONS IS REVIEWED BELOW
Previously Howard and co-workers had demonstrated in cells carrying CFTR nonsense mutations, that gentamicin induced a dose-dependent increase in expression of full-length CFTR. (see above under Howard et al)
Wilschanski M, Yahav Y, Yaacov Y, Blau H, Bentur L, Rivlin J. Aviram M, Bdolah-Abram T, Bebok Z, Shushi L, Kerem B, Kerem E. Gentamicin induced correction of CFTR function in patients with cystic fibrosis and CFTR stop mutations. N Eng J Med 2003; 349:1433-41. [PubMed]).
Further work from Israel on stop mutations (also Wilschanski et al, 2000 above). In a double-blind, placebo-controlled, crossover trial, patients with stop mutations in CFTR or patients homozygous for the DeltaF508 mutation received nasal gentamicin drops or placebo for two consecutive periods of 14 days. The gentamicin treatment caused a significant reduction in basal nasal potential difference in the 19 patients carrying one or two stop mutations (from -45 (+/-8) to -34 (+/-11) mV, P=0.005) and a significant response to chloride-free isoproterenol solution (from 0 (+/-3.6) to -5 (+/-2.7) mV, P<0.001). Also after gentamicin treatment, there was a significant increase in peripheral and surface staining for CFTR in the nasal epithelial cells of the patients carrying stop mutations.
So in patients with CF, who have premature stop codons, gentamicin was confirmed as causing translational “read through,” resulting in the expression of full-length CFTR protein at the apical cell membrane, and corrected towards normal the typical electrophysiological abnormalities caused by CFTR dysfunction. Subsequently, another compound, PTC 124, seemed to do the job more efficiently and went into clinical trials. Identified as PTC 124 (later Ataluren), the new chemical entity selectively induces ribosomal read through of premature but not normal termination codons.
Welch EM et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature 2007; 447:87-91 [PubMed].Further studies confirmed the the effect of PTC 124.
Kerem E et al. Effectiveness of PTC 124 treatment for cystic fibrosis caused by nonsense mutations: a prospective phase 11 trial. Lancet 2008; 372:719-727[PubMed].
Wilschanskli M et al. Chronic ataluren (124) treatment of nonsense mutation cystic fibrosis. Eur Respir J 2011; 38:59-69..
In June 2011 PTC therapeutics announced the results from a large multicentre Phase 3 study of ataluren (This section taken from http://ptct.client.share).
“This Phase 3 study, which was conducted across 11 countries, was a double-blind, placebo-controlled study comparing ataluren (n=116) to placebo (n=116) in nmCF patients. The primary endpoint, the relative change from baseline in %-predicted FEV1 at 48 weeks, showed a positive trend favoring ataluren versus placebo, and a larger effect in the patients not receiving chronic inhaled antibiotics. The effect of inhaled antibiotics was largely attributable to the use of inhaled aminoglycosides. In the intent-to-treat population, there was a 3% difference in the relative change from baseline in %-predicted FEV1 between the ataluren and placebo groups at Week 48 (-2.5% change on ataluren vs. -5.5% change on placebo; p=0.124). An analysis of the relative change from baseline in %-predicted FEV1 across all post-baseline study visits demonstrated an average difference between ataluren and placebo of 2.5% (-1.8% average change on ataluren vs. -4.3% average change on placebo; p= 0.0478) The study was stratified by age, baseline FEV1, and the use of chronic inhaled antibiotics.
A statistically significant effect (p=0.0072) was seen between treatment and use of inhaled antibiotics at baseline, indicating that inhaled antibiotics was a significant confounder of the overall results. A substantial treatment effect was seen in the patients not receiving chronic inhaled antibiotics at baseline; the Week 48 difference between the ataluren and placebo arms in FEV1 was 6.7% (-0.2% change on ataluren vs. -6.9% change on placebo). The secondary endpoint, the rate of pulmonary exacerbations (i.e., the number of pulmonary exacerbations in 48 weeks) also showed a positive trend in favor of ataluren, with the rate in the ataluren group being 23% lower than the placebo group (p=0.0992).
In the patients not receiving chronic inhaled antibiotics, the pulmonary exacerbation rate in the ataluren group was 43% lower than the rate in the placebo group. These results show a consistent treatment effect of ataluren on both pulmonary function and exacerbation rates. In Patients not also on inhaled antibiotics there was a 43% reduction in pulmonary exacerbations.
The FDA and the European Commission granted Ataluren Orphan Drug status for the treatment of nonsense mutation cystic fibrosis and nonsense mutation Duchenne and Beck.
The subsequent progress is reviewed in “Topics” section – “Drugs New and Old”. Eventually the drug did not go into production.
Yonemitsu Y, Kitson C, Ferrari S, Farley R, Griesenbach U, Judd D, Steel R, Scheid P, Zhu J, Jeffery PK, Kato A, Hasan MK, Nagai Y, Masaki I, Fukumura M, Hasegawa M, Geddes DM, Alton EW. Efficient gene transfer to airway epithelium using recombinant Sendai virus. Nat Biotech 2000; 18:970-973. [PubMed]
These authors developed recombinant Sendai virus (SeV) as a gene transfer agent that produces very efficient transfection throughout the respiratory tract of both mice and ferrets, as well as in freshly obtained human nasal epithelial cells in vitro.
Gene transfer efficiency was considerably greater than with cationic liposomes or adenovirus and even very brief contact time was sufficient to produce this effect and levels of expression were not significantly reduced by airway mucus. The authors suggested that SeV may provide a useful new vector for airway gene transfer.