
Eighties clinical


Eighties – clinical
Improvement in the treatment of respiratory infections and nutritional problems at specialist CF centres – a decade of substantial progress
(Summary of the Eighties Clinical; references for this are at the end of this summary)
References mentioned in this introduction to “Eighties Clinical” are listed at the end of the introduction. If the publication is summarised elsewhere this is indicated in the text by “below” or “above” indicating earlier or later in the main text. The length of this introduction to the Eighties is a reflection of the major advances in understanding and treatment that occured during the decade.
The Eighties was a decade of major advances in CF care, particularly in the UK where it must be admitted there were few centres of excellence at the start of the decade. Most children with CF in the UK received their care in general hospitals and it had not been of a standard as that given in some European centres, such as Copenhagen, and some of the main North American and Australian centres.
The description of the immunoreactive trypsin test for neonatal CF screening by Crossley and Elliot in New Zealand was a major advance (Crossley et al, 1979 above), although if neonatal diagnosis was not followed by expert care there was little advantage in early diagnosis. The IRT method was soon used in a number of successful neonatal CF screening programmes during the early and mid-Eighties e.g. East Anglia in the UK (Heeley et al, 1982 below), New South Wales (Wilcken et al, 1983 below; Wilcken & Chalmers, 1985 below), Colorado (Hammond et al, 1991 below) and Wisconsin (Farrell et al, 2001 below).
In 1975 in Leeds we started a small monthly CF clinic and enlisted the help of the paediatric outpatients nurse, physiotherapist and dietitian. In 1979 the disappointing results of a small nutritional survey, of our own and some colleagues’ patients (Congden et al, 1981 below) was a major factor in starting our so-called “Comprehensive CF Assessments” at St James’s University Hospital in Leeds in May 1980. The aim was to determine any other areas where our treatment was suboptimal and could be improved. As our first Comprehensive Assessments during the first year 1980 proved to be so useful for the management of our own patients, in 1981 the “Comprehensive CF Assessment” service was offered to paediatric colleagues throughout the Yorkshire Region in the North of England (a population of some 3.5 million). Many consultant paediatricians responded to this offer, usually first sending their most severely affected patients for an opinion. In consequence, our small team had a very steep learning curve in the treatment of CF during the early Eighties. Subsequently over 600 patients were referred to Leeds for one or more Comprehensive CF Assessments. They came from the city itself, from the surrounding Yorkshire Region, other parts of the UK and even some from overseas (Littlewood et al, 1984; Littlewood, 1986 below; Littlewood et al, 1988 below; Littlewood 1993 below; Cystic Fibrosis in Children and Adults. Revised edition Number 7. 2008; www.cfmedicine.com).
In 1988, at the International CF Congress in Sydney, we reported the results of Comprehensive Assessments on the first 250 patients seen at our Regional Paediatric CF Centre in Leeds between May 1980 and September 1987 (Littlewood JM, Kelleher J, Rawson I, Gilbert J, Firth J, Morton S, Wall C. Comprehensive assessment of patients at a CF centre identifies suboptimal treatment and improves management, symptoms and conditions. 10th International Cystic Fibrosis Congress, Sydney 1988 Excerpta Medica Asia Pacific Services. 89-90). Unfortunately, the picture was one of frequent suboptimal treatment as evidenced by incorrect diagnosis (3%), severely under-treated chest infection (as evidenced by a remarkable response following a course of intravenous antibiotics (30%), ineffective physiotherapy (60%) as judged by the physiotherapists, an energy intake which, on detailed dietary analysis, fell below the 120% of that recommended for age (75%), incorrect use of or use of older less efficient pancreatic supplements (40%) and subnormal fat-soluble vitamin levels (60%) (Littlewood et al, 1988 below). It was encouraging that the majority of families were willing to take simple advice, and having implemented our recommendations showed significant improvements in many areas when reassessed, on average 15 months after their first attendance. There were significant improvements in weight for age in the 0-5, 5-10 and the over 15 year olds. The initial poor condition of many of these patients reflected the unsatisfactory standard of treatment in many parts of the UK at the time.
It was disappointing that our paper reporting these findings was rejected by a number of leading journals – one editor noting that the critical tone of the paper was unacceptable! Also in the Eighties there was certainly a significant opposition to the need for Specialist CF Centres by many general paediatricians who could not appreciate that care at a CF Centre by a specialised team had more to offer than that given at their local clinic. “Any paediatrician worth his salt should be able to look after a cystic” one distinguished paediatric colleague was heard to observe!
As had been recommended by LeRoy Matthews of Cleveland as an essential component of his comprehensive treatment programme, we started data collection, so the results of interventions could be accurately assessed. The CF Foundation’s patient registry, developed by Dr Warren Warwick from 1964 onwards, had demonstrated a rise in median survival in the USA from 14 years in 1968 to 20 years in 1977 (Warwick & Pogue, 1969). Also Archie Norman in London published a number of papers between 1967 to 1975 recording the improving prognosis in the UK (Norman, 1967; George & Norman, 1969; George & Norman 1971; Robinson & Norman, 1975). In 1977 Mary Corey had started data collection at the Toronto CF clinic and at the 1980 Toronto International CF Congress she reported a median survival of Toronto patients of over 30 years, far better than reported by other North American centres (Corey, 1980); later she reported that the clinical state and survival of the patients were significantly better in Toronto than in Boston and also in the rest of Canada (Corey et al, 1988 below).
In North America it became clear that there were striking differences in the condition and survival of patients attending different, but nonetheless recognised and accredited, CF Centres. In the USA a study by Woods and Piazza reflected care throughout the Seventies at three recognised US CF Centres where median survivals were very different at 9.5, 18.1 and 22.8 years. Analysis of the management in the three centres showed differences reflecting particularly the closeness of supervision (number of clinic visits per year) and intensity of treatment (days of intravenous antibiotics) the patients received (Woods & Piazza, 1988 below). Interestingly this potentially sensitive data was never published other than in the proceedings of the 1988 Sydney CF Conference.
CF Centre care was already well established in Victoria, Australia to the extent that a paper published in 1984, reporting on patients during the Seventies and before, showed significantly better survival than in England and Wales where most patients still received their care at local hospitals (Phelan & Hey, 1984 below). These results prompted the formation of the British Paediatric Association UK Working Party on Cystic Fibrosis (WPCF) chaired by Professor John Dodge, who had already been to Australia and observed the treatment there. The purpose of the WPCF was to report on the situation regarding CF care in the UK. The WPCF instigated the UK Survey in 1982, supervised by John Dodge, to determine the situation in the UK at that time.
There were some 3870 patients in the UK, many saw only their local paediatrician and less than half (46.5%) attended one of the 16 clinics in the UK treating more than 50 patients. The Working Party’s eventual recommendations regarding CF Centre care were that every person with CF should have some contact with a Specialist CF Centre either by full attendance or, in the case of children via some form of shared care (UK CF Working Party Report, 1982; Dodge et al, 1988 below). It is surprising that these findings and recommendations were at first rejected (but fortunately later reluctantly accepted) by the general paediatricians on the Council of the British Paediatric Association.
Fortunately, despite the attitude of a few general paediatricians, the UK CF Research Trust, under the guidance of the then Director, Ron Tucker, gradually financed the appointment of an increasing number of doctors (CF Research Fellows), CF Nurses, Physiotherapists, Dietitians and Social Workers in the large UK hospitals where a few senior paediatricians were attempting to develop CF Centres – these appointments were absolutely essential and provided the key members of staff for the new CF Centres. Drs Leroy Matthews and Carl Doershuk, in the USA and Professor Rossi and others in Europe, had recognised the value of a specialist team approach in the Sixties. Unfortunately, it took well over 20 years for some paediatricians in the UK and elsewhere to appreciate and act on this.
During the Eighties in the UK there were major advances, almost amounting to a revolution, in the clinical care of people with cystic fibrosis. There was a gradual increase in the use of nebulised anti-Pseudomonal antibiotics for patients chronically infected with Pseudomonas aeruginosa following Margaret Hodson’s landmark paper on the use of nebulised gentamicin and carbenicillin in stabilising the condition of adults chronically infected with P. aeruginosa (Hodson et al, 1981 below). This was a major advance for these patients despite some initial reservations regarding the potential development of bacterial resistance to aminoglycosides. Here the UK was in advance of the North American clinics that were late to introduce the use of nebulised antibiotics – in fact even in 1986 a major CF centre in North America, Toronto, advised against their use (MacLusky et al, 1986 below).
In Leeds we were also worried about antibiotic resistance, but also tired of accepting that initial and almost inevitable colonisation of a CF patient with P. aeruginosa would inevitably progress to chronic infection and waiting for this to occur before starting IV antibiotic treatment. Therefore we revived the use of nebulised colomycin, an old antibiotic little used since the Sixties. Colomycin had fallen out of use but it was very active against Pseudomonas and after discussions with the manufacturers, it seemed safe to use nebulised colomycin and I decided to try to eradicate early Pseudomonas infection from the airways when first isolated before it became established. To our surprise, and against current teaching at the time, a modest half mega unit of colomycin nebulised twice daily did eradicate early P. aeruginosa infection thus delaying or preventing chronic P. aeruginosa infection (Littlewood et al, 1985 below). Subsequent clinical progress, cultures and antibody studies confirmed that eradication had taken place (Brett et al, 1986; Brett et al, 1988; Brett et al, 1992; Pond et al, 1994). The feasibility of early eradication was later confirmed in a controlled trial from Denmark using nebulised colomycin and oral ciprofloxacin (Valerius et al, 1991 below) and subsequently by a number of other studies; so early eradication of P. aeruginosa gradually became established practice in Europe – but surprisingly not until well into the Millennium in North America. Eradication of early Pseudomonas infection from the early Eighties had an obvious effect on the prevalence of chronic P. aeruginosa infection in those Centres where it was adopted e.g. it has now fallen to below 4% in children in both the Leeds (Lee et al, 2004a below), Copenhagen (Frederiksen et al, 1997 below; Frederiksen et al, 1999 below) and one Belgian CF centre where early eradication has been accepted policy since the early Nineties (Lebecque et al, 2007 below).
The earlier, more frequent and more “professional” use of intravenous antibiotics at all stages of infection (Rabin et al, 1980 below) was a major development e.g. where oral treatment had failed to eradicate a recognised pathogen or reverse a new symptom (usually a new cough) even though the patient was not ill. Choosing two antibiotics with the help of expert microbiological support, ensuring adequate blood levels of aminoglycosides and allowing for the altered pharmacokinetics of antibiotics in people with CF, all became routine in most CF Centres during the Eighties. Intensive courses of intravenous antibiotics became routine treatment for exacerbations of the chest infection (Conway et al, 1985 below) or as regular three-monthly courses in patients who were chronically infected with P. aeruginosa (Jensen et al, 1989 below). New anti-Pseudomonal antibiotics became available giving clinicians wider choice (azlocillin 1980, piperacillin 1982, netilmicin 1982, ceftazidime 1983, aztreonam 1986, and oral ciprofloxacin 1986). Improved delivery systems and intravenous access e.g. totally implantable venous access devices (Cassey et al, 1988 below; Essex-Cater et al, 1989 below), EMLA local anaesthetic cream to apply before venepuncture (which many children would rate as one of the more important advances of the decade!), butterfly cannulas, long lines, and constant intravenous infusion pumps and devices capable of delivering antibiotics and small quantities of fluid over many hours and days to maintain the patency of the vein in small children, all facilitated programmes of more aggressive intravenous antibiotic therapy. The increasing reliance on and more frequent use of intravenous antibiotics resulted in an increasing pressure on hospital beds and also the families’ domestic arrangements; so the use of home intravenous antibiotics supervised by more specialised CF staff – usually CF Nurse Specialists – gradually became used in most CF centres (Rucker & Harrison, 1974 above; Winter et al, 1984 below; Gilbert et al, 1988 below; Stern, 2001 below).
A major advance, for those who had reached the end stages of their disease in the UK, was the successful introduction of heart-lung transplantation in 1985 in the UK by Mr Magdi Yacoub at Harefields Hospital, London (Yacoub et al, 1990 below; Scott et al, 1988 below). The possibility of successful treatment in what had been previously the terminal stages of the condition had a major influence on both prognosis and the treatment of severely affected individuals. The first results of heart-lung transplantations were quite remarkable and were related both to surgical skills, concentrated medical expertise in assessment and after care and also to more successful immunosuppressive therapy to prevent rejection of the transplanted organs. Most transplant centres now report over 50% of treated patients surviving more than 5 years. Later double lung transplants became more popular (Pasque et al, 1990 below) a are now the favoured procedure. Living donor lung transplants have proved successful in some centres (Cohen & Starnes, 2001 below); at one centre in San Diego one, three and five year survivals of 70%, 54% and 45% have been reported (Starnes et al, 2004).
Liver transplantation has been used successfully in a few patients with CF and the results are surprisingly good. Respiratory function, far from deteriorating as a result of the long operation has improved in some patients (Mieles et al, 1989 below; Noble-Jamieson et al, 1994). Later successful heart-lung-liver transplantations (Noble-Jamieson et al, 1996) and lung-liver transplantations were performed (Couetil et al, 1995; Couetil et al, 1997).
Various new devices and techniques for physiotherapy of CF were described and evaluated during the Eighties (Pryor et al, 1979 below; Webber, 1986; Tyrell et al, 1986 below; Webber, 1990 below). This resulted in an increasing proportion of people with CF received effective treatment from physiotherapists who were experienced in cystic fibrosis. Many parents told me they found the hour they spent with the physiotherapist to be one of the most valuable parts of the Comprehensive Assessment at our Leeds CF Centre (Morton et al, 1988; Worthington & Kelman, 1996). Subsequently increasing attention has been paid to the value of exercise (Webb et al, 1995 below).
Also, particularly in the USA, the therapy Vest has proved very popular with many patients (Hansen & Warwick, 1990; Arens et al, 1994; Langenderfer, 1998); rather surprisingly, it is rarely used in the UK, although the cost of the device (£10,000) is undoubtedly one factor. It has been suggested that the widespread introduction of the physiotherapy vest in the USA was encouraged by the medical insurers as being more economical than routine physiotherapy. This is one of a number of interesting differences between N. American and European practice – another and more important difference being the delay in the routine use of nebulised antibiotics to eradicate early Pseudomonas aeruginosa infection.
The nutritional state of many patients continued to improve largely due to the increasing involvement of dietitians as part of most CF teams allowing accurate identification and then appropriate correction of the inadequate energy intake by individual advice and resumption of a normal or even high fat intake (MacDonald, 1984; Littlewood, 1986 below; Littlewood & MacDonald, 1987). A normal fat intake was made possible by the availability from the early Eighties of the new acid resistant enzymes, first Pancrease and then Creon. These were obviously more effective than the older unprotected preparations (Holsclaw & Keith, 1980 below; Beverley et al, 1987 below). Meyer’s studies in the late Eighties led to the introduction of the micro-sphere preparations (Myer et al, 1988). Undoubtedly these new acid resistant enzymes were one of the major advances in treatment during the Eighties and improved not only the nutrition but also the lives of the many patients who had previously had uncontrolled and severely handicapping unpleasant bowel symptoms.
For the more severely affected patients enteral feeding, first by the nasogastric route (Bradley et al, 1979 below) and then by gastrostomy or parenteral feeding (Shepherd et al, 1980 below; Levy et al, 1985 below; Shepherd et al, 1986 below) allowed nutritional rehabilitation even of those with severe malnutrition and permitted a reasonable nutritional state to be maintained even in many of the most severely affected patients e.g. those awaiting heart-lung transplantations.
Fat-soluble vitamin deficiencies were identified and re-identified and corrected by appropriate doses of suitable supplements (Sitrin et al, 1987; Kelleher et al, 1987 below; Rayner et al, 1989 below; Stamp & Geddes, 1993).
In 1989 Dr Carla Colombo of Milan first reported the beneficial effect of regular ursodeoxycholic acid treatment in improving CF related liver disease. This was an exciting prospect for, up to that time, there was no specific treatment for those with liver involvement (Colombo et al, 1990 below). Subsequent experience has confirmed the importance of this treatment particularly if started in the early stages of liver involvement. However, discussion as to its ultimate value has continued.
During the decade, as the population of people with CF in the UK increased as a result of these improvements in treatment, paediatric CF Centres developed in most large cities. Towards the end of the Eighties more CF Centres for Adults were started to treat the increasing number of people with CF who were now reaching adulthood (Conway & Littlewood, 1990; Conway, 1998 below). Also arrangements for transition from paediatrics to the adult centres received, and continues to receive, increasing attention.
References for this Summary to “Eighties Clinical”.
If a paper is summarised elsewhere in the text the reference is followed by “below” or “above”
Arens R, Gozal D, Omlin KJ, Vega J, Boyd KP, Keens TG, et al. Comparison of high frequency chest compression and conventional physiotherapy in hospitalized patients with cystic fibrosis. Am J Res Crit Care Med 1994; 150:1154-1157. [PubMed]
Brett MM, Ghoneim ATM, Littlewood JM. Prediction of diagnosis of early Pseudomonas aeruginosa infection in cystic fibrosis: a follow up study. J Clin Microbiol 1988; 26:1565-1570. [PubMed]
Brett MM, Ghoneim ATM, Littlewood JM. Serum IgG antibodies to Pseudomonas aeruginosa in cystic fibrosis. Arch Dis Child 1986; 61:1114-1120. [PubMed]
Brett MM, Simmonds EJ, Ghoneim ATM, Littlewood JM. The value of serum IgG titres against Pseudomonas aeruginosa antibodies in the management of early Pseudomonal infection in cystic fibrosis. Arch Dis Child 1992; 67:1086-1088. [PubMed]
British Paediatric Association UK Working Party on Cystic Fibrosis Report, 1982.
Cassey J, Ford WD, O’Brien L, Martin AJ. Totally implantable venous access in children with cystic fibrosis. Clin Pediatr 1988; 27:91-95. [PubMed]
Conway SP, Littlewood JM. The provision for adults at the Leeds Regional CF Unit. Association of CF Adults Newsletter. December 1990;4-5.
Corey ML. Longitudinal studies in cystic fibrosis. In: Perspectives in Cystic Fibrosis. Proc. 8th International Cystic Fibrosis Congress, Ed: Sturgess JM. Toronto: Canada, 1980:246-255.
Dodge JA, Goodall J, Geddes D, Littlewood JM, Mearns M, Owen JR, et al. Cystic fibrosis in the United Kingdom 1977-85: an improving picture. BMJ 1988; 297:1599-1602.
George L, Norman AP. Life tables for cystic fibrosis. Arch Dis Child 1971; 46:139-143. [PubMed]
George L, Norman AP. Life tables for cystic fibrosis. BMJ 1969; 3 (672);718. [PubMed]
Hansen LG, Warwick WJ. High frequency chest compression system to aid in clearance of mucus from the lung. Biomed Instrument Technol 1990; 24:289-294. [PubMed]
Jensen T, Pedersen SS, Høiby N, Flensborg EW. Use of antibiotics in cystic fibrosis. The Danish Approach. In: Pseudomonas aeruginosa infection. Antibiot Chemother 1989; 42:237-246.
Kelleher J, Miller MG, Littlewood JM, McDonald AM, Losowsky MS. The clinical effect of vitamin E depletion in cystic fibrosis. Int J Vit Nutr Res 1987; 576:253-259. [PubMed]
Littlewood AE. Cystic Fibrosis database system. In. Benefits of using clinical information. NHS Information Management Group. 1997:19-22.
Littlewood JM, Kelleher J, Losowsky MS, Page R, Crollick AJ, Miller MG, et al. Comprehensive clinical and laboratory measurements in cystic fibrosis. In: Lawson D, editor. Cystic Fibrosis: Horizons. 9th International Cystic Fibrosis Congress, Brighton. Chichester: John Wiley, 1984:266.
Littlewood JM, MacDonald A. Rationale of modern dietary recommendations in cystic fibrosis. J R Soc Med 1987; 80 (Suppl 15): s16-s24.
MacDonald A. High moderate or low fat diets for cystic fibrosis? In Cystic Fibrosis: Horizons. Ed: Lawson D. 9th International CF Congress, Brighton. Chichester, John Wiley & Sons. 1984; 395.
Miller MG, Littlewood JM. Storage, retrieval and comparison of data. The use of a microcomputer at St James’s Tertiary Referral Centre. In: Cystic Fibrosis: Horizons. Ed. Lawson D. 9th International CF Congress, Brighton. Chichester, John Wiley & Sons, 1984:263.
Langenderfer B. Alternatives to percussion and postural drainage. A review of mucus clearance therapies: percussion and postural drainage, autogenic drainage, positive expiratory pressure, flutter valve, intrapulmonary percussive ventilation, and high-frequency chest compression with the ThAIRapy Vest. J Cardiopulm Rehabil 1998; 18:283-289. [PubMed]
Morton S, Gilbert J, Littlewood JM. Physical therapy regimens of 100 consecutive patients attending a regional cystic fibrosis unit. Scand J Gastroenterol 1988; 23 (Suppl 143):110-113. [PubMed]
Meyer JH, Porter-Fink V, Elashoff J, Dressman J, Amidon GL. Human post-cibal gastric emptying of 1-3 millimetre spheres. Gastroenterol 1988; 94:1315-1325. [PubMed]
Norman AP. A life table for cystic fibrosis. Bibliotheca Paediatrica 1967; 86:368-371.
Pond MN, Carr I, Littlewood JM. A longitudinal study of anti-Pseudomonas aeruginosa ELISA in young adults with cystic fibrosis.19th European Cystic Fibrosis Conference, Paris 1994:24.
Robinson MJ, Norman AP. Life tables for cystic fibrosis. Arch Dis Child 1975; 50:962-965[PubMed]1220613
Stamp TC, Geddes DM. Osteoporosis and cystic fibrosis. Thorax 1993; 48:585-586. [PubMed]
Starnes VA, Bowdish ME, Woo MS, et al. A decade of living lobar lung transplantation: recipient outcomes. J Thorac Cardiovasc Surg 2004; 127:114-122. [PubMed]
Stern RC. Intravenous treatment: where we are and how we got there. In: Cystic fibrosis in the Twentieth Century. C F Doershuk. Editor. Ohio. AM Publishing Ltd 2001:93-111.
Warwick W, Pogue RE. Computer studies in cystic fibrosis. In: Proceedings of 5th international Cystic Fibrosis Conference, Cambridge 1969. Lawson D, editor. London: Cystic Fibrosis Research Trust, 320-330.
Webber BA. Hofmeyer JL, Moran MDL, Hodson ME. Effects of postural drainage incorporating forced expiration technique, on pulmonary function in cystic fibrosis. Br J Dis Chest 1986; 80:353-359. [PubMed]
Webber BA. The active cycle of breathing exercises. Cystic Fibrosis News 1990; Aug/Sept: 110-111.
Winter RJD, George RJ, Deacock SJ, Shee CD, Geddes DM. Self-administered home intravenous antibiotic therapy in bronchiectasis and adult cystic fibrosis. Lancet 1984; i:1338-1339. [PubMed]
Worthington D, Kelman BA. Current physiotherapy practice of new referrals to a regional paediatric cystic fibrosis service. Physiotherapy 1996; 82:253-257.
Eighties clinical references in chronological order
1980 Boxerbaum B. Isolation of rapidly growing mycobacteria in patients with cystic fibrosis. J Pediatr 1980; 96:689-691. [PubMe
The first report of non-tuberculous mycobacteria in patients with cystic fibrosis – an infection that would be increasingly important.
1980 Wood RE, Sherman JM. Pediatric flexible bronchoscopy. Ann Otol Rhinol Laryngol 1980; 89:414-416. [PubMed]

Fig 1. Robert Wood
The first report of the use of the paediatric flexible fibreoptic bronchoscope in children by Robert Wood (figure 1) who pioneered the technique. He described a prototype flexible paediatric bronchoscope that he had used to perform both diagnostic and therapeutic procedures on pediatric patients ranging from infants of 840 g to children aged 14 years. Flexible bronchoscopy, with appropriate instrumentation and careful attention to physiological requirements of the patient, was found to be safe and effective in young patients. Wood correctly forecast that with this instrument, the indications for bronchoscopy in children would be considerably expanded.
(Also there is an excellent review of flexible bronchoscopy by Wood RE & Postma D. J Pediatr 1988; 112:1-6 [PubMed]; also Wood RE. “Pediatric flexible bronchoscopy: The inside story”. In: Doershuk CF, (Ed.). Cystic Fibrosis in the Twentieth Century. Cleveland: AM Publishing Ltd, 2001:112-119.).
The indications and availability of paediatric flexible bronchoscopy continued to expand and it has even been suggested that every infant identified by newborn screening should be bronchoscoped to obtain cultures of the lower airways (Hilliard TN et al. Arch Dis Child 2007; 92:898-899. [PubMed]). However, provisional data from a more recent major study from Australia by Claire Wainwright and colleagues would not support this suggestion, the information from regular bronchial lavages leading to no better outcome than from routine therapy (Wainwright CE et al. Effect of bronchoalveolar lavage-directed therapy on Pseudomonas aeruginosa infection and structural lung injury in children with cystic fibrosis: a randomized trial. JAMA 2011; 306:163-171). [PubMed]
1980 Cohen LF, Di Sant’Agnese PA, Friedlander J. Cystic fibrosis and pregnancy: a national survey. Lancet 1980; ii: 842-844. [PubMed]
The first large national survey of pregnancies in women with CF attending 119 CF centres in the U.S.A. and Canada, reporting 129 pregnancies in 100 women. Ninety seven pregnancies were completed resulting in 86 viable infants, only one of whom had CF. Shortened gestation and increased maternal and perinatal mortality were related to severe maternal pulmonary infection. There were no congenital anomalies in this series in spite of frequent use of antibiotics by these mothers.
The first report of a pregnancy in a woman with CF was by Siegel & Siegel (1960 above) and the patient died six weeks after the birth. Even in 1980 pregnancy was still a hazardous undertaking for women with CF and the authors of this survey advised that unless the clinical condition was good, pregnancy should be avoided in women with cystic fibrosis. Fortunately, although there were still problems, the outlook continued to improve (Gilljam M, et al. Chest 2000; 118:85-91. [PubMed] below; Edenborough FP, et al. Brit J Obstet Gynaec 2000; 107:254-261. [PubMed] below).
1980 Mearns MB. Natural history of pulmonary infection in cystic fibrosis. In: Perspectives in Cystic Fibrosis. Ed: Sturgess JM. Toronto 1980; 325-334.
An interesting review, by one of the UK’s few paediatric CF experts, Margaret Mearns, describing the gradual transition from Staphylococcus aureus infection to Pseudomonas aeruginosa as the main infecting organism. This was considered to be due to an increasing use of antibiotic therapy and perhaps changes in the bacteria in the environment.
1980 Holsclaw DS, Keith H. Long-term benefits of pH sensitive enteric coated enzymes in CF. Perspectives in Cystic Fibrosis. Proc. 8th International Cystic Fibrosis Congress Toronto 1980. 19a.
One of the first reports of the new acid resistant pancreatic enzyme – Pancrease. Previously discussed at the N. American Cystic Fibrosis Club by Khaw et al, 1977, Suskind et al, 1979 and Weber et al, 1979 (all above). This report, presented as a poster, provided details of longer term treatment to that already reported at the 1979 Cystic Fibrosis Club.
Twenty patients with CF were followed for 14 months. Urine uric acid decreased from 850 to 550 mg/day, diets broadened in terms of fat content, nutritional state improved and gastrointestinal symptoms diminished on between nine and 11 Pancrease capsules per day.
The obvious superiority over previous enzyme preparations was repeatedly confirmed in subsequent controlled trials (Mischler et al, 1982; Gow et al, 1981; Beverley et al, 1987 – all below). Over 90% of the enzymatic activity of the older unprotected preparations was destroyed by the acid in the stomach; the new preparations passed through the stomach before releasing their enzyme activity (figure 1a).

Fig 1a. Intact Pancrease microspheres enzymes in duodenal fluid
So these new acid resistant microspheres (figure 2) were undoubtedly one of the major advances of the decade

Fig 2.Microsphere enzymes Creon and Pancrease
permitting most people with CF to eat a normal amount of fat.
At the time of these initial reports it was hard to imagine the profound beneficial effect the enzymes were to have on the patients’ ability to tolerate fat, improve their energy intake and maintain a reasonable nutritional state and, not least, improve their quality of life.
However, when one observed the effect on people with CF their superiority over the older preparations was obvious.
One 16 year old girl wept as she recounted to me how her life, previously dominated by her very abnormal bowel habit, had been totally transformed by the new enzymes as she was now able to take part in normal social activities with her friends. (also Weber et al, 1979 above; Khaw et al. 1977&1979 above; Holsclaw DS & Keith H, 1980 above; Mischler et al, 1982 below; Beverley et al, 1987 below).
1980 Rabin HR, Harley FL, Bryan LE, Elfring GL. Evaluation of high dose tobramycin and ticarcillin treatment protocol in cystic fibrosis based on improved susceptibility criteria and antibiotic pharmacokinetics. In Perspectives in Cystic Fibrosis. Ed: Sturgess JM. 8th International Cystic Fibrosis Congress, Toronto, Canada 1980; 370-375.[Conference Proceedings]
This paper, although not widely quoted, had a major influence on our approach to intravenous antibiotic treatment – indeed to our whole more intensive approach to treating people with cystic fibrosis.

Fig 3. Harvey Rabin
At the 1980 Toronto meeting I was very impressed by the team approach and the significant involvement of all the members of the CF team and their obvious professional approach. With Archie Norman and his wife, who were also attending the meeting, I had an opportunity to visit Henry Levison, the paediatrician in charge of the CF Unit at the Hospital for Sick Children, Toronto.
The whole format of the conference at the Royal York Hotel, Toronto was quite new to me.
This paper by Harvey Rabin (Fig. 3) and colleagues on intravenous antibiotic therapy was typical of the more professional approach to IV antibiotic therapy appropriate for CF which had been developed during the Seventies in some of the larger North American CF Centres (Stern RC. Intravenous treatment: Where we are and how we got there. In: Doershuk CF, (Ed.). Cystic fibrosis in the Twentieth Century. Cleveland: AM Publishing Ltd, 2001:93-111).
The frequent use of intravenous antibiotics for children with CF was not usual practice in most of the UK hospitals in 1980 where the majority of children with CF were still treated by their local general paediatricians. Certainly this meeting at the Royal York Hotel, Toronto was the start of an increasingly serious involvement in Leeds with the whole field of cystic fibrosis – both research and clinical care. In retrospect, it was undoubtedly the centre/team approach first started by Leroy Matthews in Cleveland and the “Not so fatal disease” Toronto approach of Douglas Crozier (Crozier, 1974 above), that were the central lessons which I took back to Leeds in 1980.
Dr Harvey R Rabin (fig.3) was in 1980 at the time of this presentation Professor in the Department of Microbiology, Immunology and Infectious Diseases, University of Calgary. He has published extensively on the microbiological and other aspects of cystic fibrosis during a long and distinguished career; he received an AMA Award for Distinguished Service.
1980 Shepherd R, Cooksley WGF, Cooke WD. Improved growth and clinical, nutritional and respiratory changes in response to nutritional therapy in cystic fibrosis. J Pediatr 1980; 7:351-357. [PubMed]

Fig 4. Ross Shepherd
One of the early papers by Ross Shepherd (fig. 4) of Brisbane on the use of parenteral nutrition in cystic fibrosis.
Twelve malnourished children with CF were studied from six months before to six months after a period of supplemental parenteral nutrition. After parenteral nutritional therapy, providing a balanced consistent hyper caloric intake for 21 days, catch-up weight gain occurred by one month and continued at six months; catch-up in linear growth was observed by three months and continued at six months; also there were fewer respiratory infections.
Here was further evidence, from Ross Shepherd, of the impressive benefits of this new aggressive nutritional intervention with parenteral feeds. Subsequently there were further studies from Brisbane on nutritional rehabilitation.
1980 Matthews WJ Jr, Williams M, Oliphint B, Geha R, Colten HR. Hypogammaglobulinemia in patients with cystic fibrosis. N Eng J Med 1980; 302:245-249. [PubMed]
Serum immunoglobulins were measured in 419 people with cystic fibrosis. Twenty-two per cent of the 154 patients less than 10 years old had hypogammaglobulinemia-G, whereas the older patients had normal or elevated serum immunoglobulins. The patients with hypogammaglobulinemia had significantly less severe lung disease than did age-matched patients with normal or elevated IgG levels. The authors suggested that the progression of lung disease may be due in part to a hyper-immune response not present in those with the hypogammaglobulinemia.
Subsequent studies of immunoglobulins (Garside JP et al. Pediatr Pulmonol 2004; 39:135-140) also showed a number of patients with low immunoglobulins and more with low subgroups – particularly low IgG2. As in previous studies the minority of patients with high IgG levels were in worse clinical condition. A follow up of these patients (Garside et al. 2007; 42:125-130) showed there was a reduction in the prevalence of low levels of IgG2 from 29% to 10% over the 2-year period. Low levels of IgG2 were not associated with any decline in clinical well-being. Again the children with high levels of IgG2 had worse lung function, worse clinical and chest X-ray scores and higher levels of P. aeruginosa infection. Children with low IgG2 levels were not worse clinically compared to those with normal or high IgG2 levels. There was no subsequent explanations for these findings.
1980 Brueton MJ, Omerod LP, Shah KJ, Anderson CM. Allergic bronchopulmonary aspergillosis complicating cystic fibrosis in childhood. Arch Dis Child 1980; 55:348-353. [PubMed]
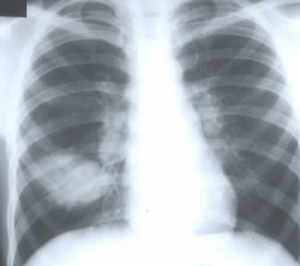
Fig 6. Allergic broncho pulmonary aspergillosis
This report in the leading widely read UK paediatric journal, by Martin Brueton (fig. 5) when he was working in Professor Charlotte Anderson’s department in Birmingham. It was important as it brought to the attention of general paediatricians the complication of allergic bronchopulmonary aspergillosis (ABPA) in children with CF. The problem had been described previously in older patients with CF (Mearns et al, 1965)

Fig 5. Martin Brueton
This paper certainly made us more aware of ABPA – the condition for which steroids and a not antibiotics were required. The complication came to be suspected in the presence of increasing asthmatic symptoms accompanied by major chest X-ray changes ranging from total or lobar lung collapse to extensive but changing areas of consolidation (fig. 6) associated with eosinophilia, positive skin tests and positive serum precipitins for Aspergillus. There was not the expected clinical response to intravenous antibiotics. Sometimes the extent of the often major segmental x-ray changes was greater than and quite out of keeping with the outward clinical disturbance. Large doses of oral steroids were recommended and are still used but few would now agree with the authors of the present article that there was no place for anti-fungal agents, for these now have an increasing place in treatment to reduce the load of Aspergillus antigen present. As more intravenous antibiotics were used during the Eighties the incidence of ABPA increased – this was the experience both in Copenhagen and Leeds.
1981 Martin AJ, Smalley CA, George RH, Healing DE, Anderson CM. Gentamicin and tobramycin compared in the treatment of mucoid Pseudomonas lung infections in cystic fibrosis. Arch Dis Child 1980; 55:604 – 607. [PubMed]

Fig. 6a Charlotte Anderson Univ. Birmingham
A small, straightforward and practically useful clinical comparison from Charlotte Anderson’s (6a) unit Birmingham showing the two intravenous antibiotics, when combined with carbenicillin, gave similar results when used for treating respiratory exacerbations in cystic fibrosis. Subsequently gentamicin was shown to be less effective in vitro against Pseudomonas and was definitely the more nephrotoxic and ototoxic of these two aminoglycosides. Therefore tobramycin became the preferred aminoglycoside in most CF clinics.
This was a useful review of the use of intravenous aminoglycosides in CF at the time – then a relatively new practice to many UK paediatricians. It is perhaps a poor reflection on our UK National Health Service that some CF Centres in the UK were still using the more toxic IV gentamicin in 2008 apparently as their laboratory would only estimate blood levels of gentamicin and not tobramycin. A UK survey of renal failure in CF in 2007 showed virtually all those with episodes of acute renal failure had received gentamicin rather than tobramycin (Bertenshaw et al, 2007 below).
1981 Schiotz PO, Hoiby N, Flensborg EW. Cystic fibrosis in Denmark. In: Warwick WJ. Ed: 100 years of Cystic Fibrosis. Minnesota 1981:141-146.

Fig. 7. The Rigshospitalet in Copenhagen where the CF unit is based (from Wikipedia website)
A report describing the Copenhagen regimen including the practice of giving 3-monthly courses of IV anti-Pseudomonal antibiotics to patients who were chronically infected. This practice was not accepted by most CF clinicians but at that time there was a great discrepancy between the CF care and results in most parts of the UK and the intensive approach to care in the Copenhagen CF centre (fig. 7) where a major influence was that of Niels Hoiby. The regimen was introduced to anticipate and prevent the deterioration of lung function after a course of IV antibiotics (also Szaff et al. 1983 below).
It should be noted that regular inhaled antibiotics were not used in Copenhagen at this time to stabilise the chest infection between the courses of IV antibiotics. The use of regular inhaled antibiotics, was largely influenced by Margaret Hodson’s 1981 paper (Hodson et al, 1981 below) and had a favourable influence on stabilising the Pseudomonas chest infection making 3 monthly courses of IV antibiotics less necessary in some patients. In Copenhagen our report on inhaled colomycin in 1985 (Littlewood et al, 1985 below) prompted their use of inhaled colomycin in Copenhagen from 1987 for chronically infected patients (Jensen et al, 1987, below). Most UK CF Centres had a flexible policy using IV antibiotics early but as and when required rather than on a rigid 3-monthly basis.
1981 Norman AP. Cystic Fibrosis and normality. In: Warwick WJ. 1000 Years of Cystic Fibrosis. Minnesota 1981:84-89.

Archie Norman MBE
A paper by Archie Norman given to leaders of CF research and clinical care who were invited to attended this meeting in Minnesota in 1981 organised by Dr. Warren Warwick. Dr. Archie Norman pleads for a change in attitude. “It was time we stopped talking in terms of the most lethal genetically determined disease” and always discussing topics such as the “problems of individuals expected to die in early childhood”.
1981 Webster Hl, Barlow WK. New approach to cystic fibrosis diagnosis by use of an improved sweat-induction/collection system and osmometry. Clin Chem 1981; 27:385-387.[PubMed]
First use of osmometry of the sweat to distinguish people with CF from controls. The method was validated in a number of subsequent studies (Kirk JM et al. 1983 below) but never became widely adopted or and did not replace the standard sweat test.
1981 Congden PJ, Bruce G, Rothburn MM, Clarke PCN, Littlewood JM, Kelleher J, Losowsky MS. Vitamin status in treated patients with cystic fibrosis. Arch Dis Child 1981; 56:708-714.[PubMed]
Fig 8a. Peter Congdon
This was our first nutritional research study from Seacroft Hospital and St James University Hospital, Leeds collaborating with the University Department of Medicine at St James’s. I presented the data at the 1980 Toronto CF Conference. It was the first of many studies on the nutritional
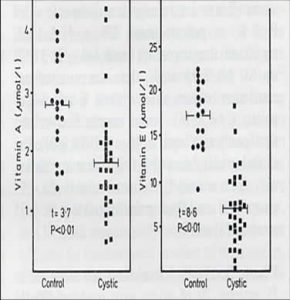
Fig 8. Vitamin A and E results from Congden et al paper .With permission of BMJ Publishing Group
and gastrointestinal aspects of CF carried out in collaboration with Dr. Jerry Kelleher and other members of Professor Monty Losowsky’s Department of Medicine at St James’s in Leeds.
This study was coordinated by the late Dr. Peter Congdon (Fig.8a) (correctly spelt Congdon in all future publications). Many of our patients, and those of some of our paediatric colleagues who were also included, had unexpectedly low fat-soluble vitamin levels and suboptimal control of intestinal absorption despite what we considered to be adequate enzyme and vitamin supplements; but the water soluble vitamin levels were satisfactory.
These disappointing results (fig 8) , reflecting our suboptimal treatment in the late Seventies, prompted us to start Annual Comprehensive CF Assessments in May 1980 along the lines suggested by Crozier in 1974; he stated that “success of treatment will depend on a complete assessment of the patient and then continuing attempts to obtain normal bodily function and maintain it”.
The late Dr Peter Congdon (fig. 8a) was our Senior Registrar at the time of this study. He played a major part in coordinating this and a number of other studies during his time with us at St James’s. He also shouldered much of the work of the large neonatal unit at St James’s. Peter was eventually appointed consultant neonatologist at the Leeds General Infirmary where he ran a very successful regional neonatal intensive care service until his untimely death.
Our so-called “Comprehensive CF Assessments” proved so useful in identifying areas where we could improve treatment of our own patients, that in 1981 I offered the service to paediatric colleagues in the Yorkshire Region for their patients. It was through this offer, to accept patients for assessment and advice, that the Leeds Regional CF Service developed.
1981 Szaff M, Hoiby N. Antibiotic treatment of Staphylococcus aureus infection in cystic fibrosis. Monogr Paediatr 1981; 14:108-114. [PubMed]
This was a very helpful practical account of the Danish regimen for treating S. aureus based on 15 years experience with 209 patients. If the organism was cultured from the airways of a person with CF “anti-Staphylococcal therapy was given whether there were clinical symptoms or not – usually courses of oxacillin and dioxacillin with fusidic acid for 14 days. Chronic infections were given long term treatment for one to three months”. The authors noted no increase in P. aeruginosa from eradicating S. aureus and only 10% of patients in the Danish clinic had developed chronic S. aureus infection – in contrast to the usual 50% prevalence in many CF clinics. Each patient required an average of two courses of anti-staphylococcal antibiotics per year to achieve these results. Only a small increase in the number of S. aureus precipitins was observed. In the UK David Lawson in London had also prevented the development of precipitins by long term cloxacillin treatment (Lawson D. Arch Dis Child 1976; 51:890-891 above).
In our expanding CF clinic in Leeds, we found this paper from Copenhagen to be very useful as it gave clear guidance on antibiotics and dosages and also showed that chronic Staphylococcal infection could be prevented by close frequent microbiological monitoring and early treatment with short courses of antibiotics as an alternative to David Lawson’s long term cloxacillin – this was useful as some doctors and families were hesitant about long term antibiotic treatment. However, we eventually opted for long term flucloxacillin from diagnosis for all patients as recommended by David Lawson and this is still the Leeds policy resulting in a low overall prevalence of chronic S. aureus infection (14%) compared to many centres (40-50%) and in only 8.3% (5/60) of children under 10 years of age.
It is difficult to understand how chronic infection with S. aureus, a known harmful pathogen shown by bronchial lavage studies to result in an inflammatory reaction within the airways, is allowed to remain in so many patients when chronic infection can be prevented? With regard to the influence of this treatment on the prevalence of P. aeruginosa infection, the prevalence of chronic P. aeruginosa in the Leeds clinic, where all patients are on long term flucloxacillin, is less than 4% in children under 12 years with CF (Lee et al, 2004 below). However, there has been a policy of early eradication treatment of Pseudomonas since 1984 and neonatal screening and early treatment from 1975 neither of which are present in the US centres reporting an increase in Pseudomonas with anti-Staphylococcal treatment (Stutman HR et al. J Pediatr 2002; 140:299-305. [PubMed]).
1981 Evans RT, Little AJ, Steel AE, Littlewood JM. Satisfactory screening for cystic fibrosis with the BM meconium procedure. J Clin Path 1981; 34:911-913. [PubMed]
Dr Robert Evans, at the time the consultant biochemist at St James University Hospital, Leeds , agreed to perform our BM Meconium tests in the laboratory when it became obvious the midwives on our maternity wards were far too busy looking after the mothers. Here he reported favourable results since we started BM screening in 1975. By 1981 most paediatricians had either never started or by this time rejected the BM Meconium test for neonatal CF screening, yet in one maternity hospital in East Leeds (St Mary’s) the test had become routine and we had continued to use the test from 1975. We had more success than other places with a false negative rate of only 12% – but we found, as had the people in Veneto, that the test had to be done in the laboratory and not by midwives who, it was soon realised, had their hands full looking after the mothers and babies!
The test was used continuously in East Leeds until the early 1990s when we changed to immunoreactive trypsin (now IRT/DNA/IRT) and involved the whole city. I thought continuous CF neonatal screening from 1975 was a world record; however it appears to have been just preceded by Gianni Mastella’s regional CF neonatal screening in Veneto Italy from 1974!! (Mastella et al, 1981 below)
1981 Mastella G, Borgo G, Castellani E, Ferro I, Pederzini F. Results and praxis for cystic fibrosis neonatal screening, steps in a regional program and some evaluation of the prophylactic treatment (A story of 8 year cystic fibrosis screening in Veneto). In: Warwick WJ (ed.) 1000 Years of Cystic Fibrosis. Minnesota University, 217-232.
When many people were arguing about the possible benefits of neonatal screening (as occurred in the UK until 2001 and even more recently by some) some paediatricians introduced screening as it seemed a commonsense idea to diagnose early and treat vigorously an eventually fatal condition where survival depended almost entirely on the success of the treatment!

Fig 9. Gianni Mastella
Professor Gianni Mastella ( – 2021) (fig. 9) was one such paediatrician – since 1960 he produced 177 publications – 122 on CF and has been one of the outstanding pioneers of European CF care (for which he was awarded the Rossi Medal of the European CF Society), a pioneer of neonatal CF screening and one of the senior doctors of Italian CF. Gianni Mastella died in 2021.
This report concerns five stages of the Veneto programme from 1974 to1981, screening 200,000 infants in 81 hospitals. Stages 1 to 3 used the BM test. As we also discovered in Leeds, the BM test was acceptable only if performed in the laboratory but not when performed by many different nurses. Stages 4 and 5 used an immunodiffusion technique for albumin when there were only 9.5% false negatives. Finally IRT in blood spots (as described by Crossley et al, 1979) was introduced. Nutritional parameters and clinical scores were significantly better in the screened infants.
1981 Gow R, Bradbear R, Francis P, Shepherd R. Comparative study of varying regimens to improve steatorrhoea and creatorrhoea in cystic fibrosis: effectiveness of an enteric coated preparation with and without antacids and cimetidine. Lancet 1981; 2: 1071-1074. [PubMed]
This was a useful early paper further confirming the marked superiority of the recently introduced Pancrease microsphere enzymes over conventional pancreatic enzyme preparations. Ten patients were observed during four two-week treatment periods of 1). Conventional pancreatic supplements. 2). pH sensitive enteric coated microspheres (Pancrease). 3). Enteric coated microspheres (ECMP) plus cimetidine. 4). ECMP plus antacids. Significant reductions in faecal fat, nitrogen and faecal weight occurred with the Pancrease. Additional antacids and cimetidine did not improve absorption except in those where there was still significant malabsorption when the addition of cimetidine caused significant improvement. (also Weber et al, 1979 above; Khaw et al. 1977&1979 above; Holsclaw DS & Keith H, 1980 above; Mischler et al, 1982 below; Beverley et al, 1987 below).
1981 Hodson ME, Penketh ARL, Batten JC. Aerosol carbenicillin and gentamicin treatment of Pseudomonas aeruginosa in patients with cystic fibrosis. Lancet 1981; i: 1137-1139. [PubMed]

Fig. 10 Margaret Hodson The BMJ
Another definite landmark paper from the Adult CF Unit at the Royal Brompton Hospital, London. by Professor Margaret Hodson (fig. 10). Margaret was to make many major contributions to the treatment of people with CF from her vast experience at the Brompton Hospital treating many hundreds of adult patients with CF.
This paper undoubtedly had a major favourable influence on treatment in the UK. There had been many earlier concerns about increasing the incidence of bacterial resistance from nebulised antibiotics. Nebulised penicillin had been used in the Forties when S. aureus was the main pathogen (di Sant’Agnese, et al, 1946 above). However, it was undoubtedly this present paper that revived the interest in nebulised antibiotics for patients with chronic Pseudomonas infection – particularly in the UK. The nebulised anti-Pseudomonal antibiotics obviously stabilised the condition of some patients with frequently relapsing chronic P, aeruginosa infection who were requiring increasingly frequent courses of IV antibiotics (figure 11) and represented a major milestone in treatment.
As a result of this paper nebulised anti-Pseudomonal antibiotics became widely used in the UK for patients with chronic Pseudomonas infection. In
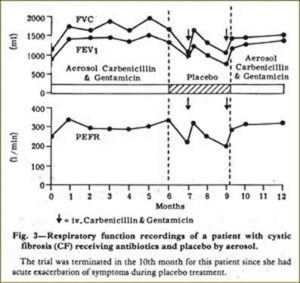
Fig 11. Chart from Hodson et al paper showing patient receiving either antibiotics or placebo
this respect the UK was well in advance of North America where, even in 1986, McLusky and colleagues from Toronto advised that, “until additional well-controlled trials were completed their routine use (of inhaled antibiotics) was not justified because of cost, potential side effects and the propensity to select resistant organisms” (McLusky et al, 1986 below).
As it turned out, both the routine use of nebulised anti-Pseudomonal antibiotics to suppress chronic infection, as recommended by Margaret Hodson in 1981 (also eventually by Ramsay BW et al, 1999) and the early use of colomycin to eradicate early Pseudomonal infection (Littlewood et al, 1985; Valerius et al, 1991) both proved to be effective, proven and eventually widely used treatments for people with CF on both sides of the Atlantic.
In 2012 Margaret Hodson was awarded a well-deserved OBE for services to medicine.
1981 Laughlin JJ, Brady MS, Eigen H. Changing feeding trends as a cause of electrolyte depletion in infants with cystic fibrosis. Pediatrics 1981; 68:203-207. [PubMed]
Five infants with CF had eight episodes of hypoelectrolytemia – six of these were not associated with high environmental temperature and were considered possibly related to the current trends in reducing the salt in infant formula feeds. The salt content of infant formula feeds had been reduced in view of the reports of hypernatraemic dehydration which occurred in some infants on formula milk. But just as the incidence of hypernatraemic dehydration in non-CF infants diminished when the salt content of infant feeds was reduced, so some infants with CF were adversely affected by the reduction in their dietary salt. Eventually salt supplements were recommended for CF infants.
1982 Heeley AF, Heeley ME, King DN, Kuzemko JA, Walsh MP. Screening for cystic fibrosis by dried blood spot trypsin assay. Arch Dis Child 1982; 57:18-21.[PubMed]

Fig 12. Anthony and Mary Heeley in 2012
The first detailed report of immunoreactive trypsin neonatal CF screening in East Anglia in the UK by Anthony Heeley the biochemist in Peterborough and his wife Mary (figure 12) and colleagues. The Heeleys were pioneers of neonatal immunoreactive trypsin CF screening in the UK.

Fig. 12a Jan Kuzemko
Dr Jan Kuzemko (1933-2019) (Fig12 a) was their paediatric colleague in Peterborough and also involved in a number of early CF publications from the UK including the use of continuous IV ceftazidime (1989 [PubMed], resistance with ceftazidime monotherapy (1988) [PubMed] and home intravenous antibiotic treatment (1988). [PubMed]
In the present study 14,000 infants were screened, 0.2% required a second test and five infants with CF were detected. A satisfactory method which has continued to the present time and resulted in a number of subsequent publications from East Anglia (Green MG et al. Cystic fibrosis identified by neonatal screening: incidence, genotype, and early natural history. Arch Dis Child 1993;68: 464-467; Green MR & Weaver LT, J R Soc Med 1994; 87 suppl 21:5-10; Weaver LT et al Arch Dis Child 1994; 70:84-89 below). This last was an important controlled trial on the effect of long term flucloxacillin during the first 2 years although its design did not reach the high stahdards some colleagues expect.
In the longer term it was unfortunate that many of these screened infants in East Anglia did not receive specialist CF centre care, as was usual at the time, and consequently many did not achieve their full health potential (see Crossley & Elliott, 1979 above in Seventies for Dr Heeley’s recent detailed comments on the early use of IRT for neonatal screening).
A 30 year follow-up of this programme was published in 2012 and Anthony and Mary Heeley were among the authors (Calvin J, Hogg SL, McShane D, McCauleySA, Illes R, Ross-Russell R, MacLean FM, Heeley ME, Heeley AF. Thirty-years of screening for cystic fibrosis in East Anglia. Arch Dis Child 2012; 97:1043-1047. [PubMed] During the 30 years 582,966 infants were screened by IRT-IRT and 147,764 by IRT-DNA-IRT (total 730,730 infants) resulting in 296 screen positive cases of CF and 29 false negatives (including 10 with meconium ileus).
1981 Travert G, Mustin C, Fernandez Y. Radioimmunoassay of trypsin in dried blood. Importance for the neonatal detection of cystic fibrosis. Nouv Presse Med 1981; 10:2093-5.[PubMed]

Fig 13. Mary & Anthony Heeley with George Travert
(First author’s translation) The demonstration of very high levels of immunoreactive trypsin in the blood of newborn infants with cystic fibrosis has provided a new way of detecting the disease soon after birth. A radioimmunoassay of trypsin in the eluate of blood dried on filter paper has now been developed. The sensitivity and accuracy of the method, as well as the good correlation observed between the values obtained and those of the conventional plasma assay, indicate that it is reliable and well adapted to the newborn. The new assay can easily be inserted into the present system of neonatal disease detection. A preliminary assessment of more than 5,000 tests enables the authors to report an early diagnosis of proven cystic fibrosis and to discuss an essential aspect of mass-detection methods: the incidence of false-positive results.
Dr George Travert (fig. 13) of Laboratoire de Biophysique, Caen was involved in IRT neonatal CF screening in Brittany from soon after the publication of the original paper by Jeanette Crossley in 1979 (above) (Travert G et al. J Genet Hum 1980; 28:141-4.) as was Anthony Heeley in the UK. He continued to make major contributions for almost 30 years to CF screening most recently as a member of the Committee on European best practice guidelines for cystic fibrosis neonatal screening (J Cyst Fibros 2009; 8: 153-173. [PubMed]). George Travert was also the principal organiser of a very successful international conference on neonatal CF screening in Caen in 1999 which was attended by virtually everyone involved in neonatal CF screening over the previous 20 years (Depistage neonatal de la mucoviscidose. Presses Universitaires de Caen, 1999).
1982 Mischler EH, Parrell S, Farrell PM, Odell GB. Comparison of effectiveness of pancreatic enzyme preparations in cystic fibrosis. Am J Dis Child 1982; 136:1060-1063. [PubMed]
One of the early trials establishing the clear superiority of the new acid-resistant pH sensitive microsphere enzymes (Pancrease) over the conventional encapsulated powders (Cotazym). Ten boys in the trial experienced significantly better nitrogen and fat absorption with the enteric-coated enzyme product Pancrease. (also Weber et al, 1979 above; Khaw et al. 1977&1979 above; Holsclaw DS & Keith H, 1980 above; Gow et al, 1981 above; Beverley et al, 1987 below).
1982 Kelly NM, Fitzgerald MX, Tempany E, O’Boyle C, Falkiner FR, Keane CT. Does Pseudomonas cross infection occur between cystic fibrosis patients? Lancet 1982; 2:688-690. [PubMed]

Fig 14. Eddie Tempany
This study from Prof. Eddie Tempany’s unit in Dublin (fig. 14) was published long before it was appreciated that Pseudomonas could to spread between patients in CF clinics and at a time when Pseudomonas positive patients mixed freely with Pseudomonas negative patients in all CF centres, clinics and socially.
Over a 12-month period respiratory Pseudomonas aeruginosa isolated from CF patients were typed by serology and pyocin production to determine whether cross-infection was occurring. Although one strain appeared in four unrelated patients, none of these patients had been in contact with each other and the strains were considered to have been acquired from the environment. However, it is relevant that each of six pairs of siblings with CF shared the same strain, but the pairs of strains were distinct from each other.
These results suggested to the authors that the general environment was the most important source of Pseudomonas strains for CF patients and that for cross-infection to occur prolonged intimate contact was required – such as living in the same household.
This was an early study on the possibility of cross infection which at the time was considered to be reassuring. However subsequently, with the advent of more sensitive genetic testing, cross infection was shown to be relatively common in CF centres and clinics, although in 1982 there were relatively few such large groups of patients with CF in the UK.
1982 British Paediatric Association (BPA) Working Party on Cystic Fibrosis was formed. Supported by the Royal College of Physicians of London (RCP) and the British Thoracic Society (BTS).
This was the start of a major change in the care of people with CF in the UK. The Phelan & Hey 1984 data was available in 1982 (Phelan & Hey, 1984 below) and prompted the formation of this British Paediatric Association Working Party on Cystic Fibrosis – funded by the UK CF Trust. Their brief was “To assess the advantages and disadvantages of regional centres for cystic fibrosis”. The members of the Working Party were John Dodge (Chairman, Paediatrician), Janet Goodall (Paediatrician), Duncan Geddes (Respiratory Physician), Jim Littlewood (Paediatrician), Margaret Mearns (Paediatrician) and George Russell (Paediatrician).
The working party initiated the UK Survey to determine the present state of the UK patients. The findings were summarised by Tony Jackson on behalf of the CF Trust (Working Party on Cystic Fibrosis. Arch Dis Child 1986; 61:724 below).
Also a fuller report eventually appeared (BMJ 1988; 297:1599-1602 below). John Dodge recalls that the report was accepted by the Royal College of Physicians and the British Thoracic Society but initially rejected by the British Paediatric Association council who were reluctant to have their patients’ care transferred to CF centres; the paediatricians required it to be “watered down” before it was eventually published!

Fig 15. Warren Warwick, John Dodge & Jean Fiegelson
Prof. John Dodge CBE ( -2022) (fig. 15) was Director of the Cardiff CF Centre before moving to Belfast as Professor of Paediatrics. He has been involved in CF for many years and published widely on the subject. He set up regional CF clinics in Northern Ireland in 1965 when a lecturer there and in Cardiff in 1971. He was Chairman of the Scientific and Medical Advisory Committee of the International Cystic Fibrosis (Mucoviscidosis) Association and has chaired CF working parties for the WHO. A major contribution was the supervision and ongoing analysis of the data collection of which started with the UK Working Party in 1982, the final analysis of which was published in 2007 (Dodge et al, Eur Respir J 2007; 29:522-526) [PubMed].
1982 Gaskin K, Gurwitz D, Durie P, Corey M, Levison H, Forstner G. Improved respiratory prognosis in patients with cystic fibrosis with normal fat absorption. J Pediatr 1982; 100:857-862. [PubMed]

Fig 16. Gordon Forstner

Fig.16a Kevin Gaskin. Author’s photo
A study carried out when Kevin Gaskin (Fig.16a) (1948-2021) from Sydney was working in Toronto. In general, patients who were pancreatic sufficient had milder clinical symptoms and a lower mean sweat chloride value (85.42+-24.43 SD meq/l) than their counterparts with steatorrhoea (102.29 +-20.4 meq/l); also their pulmonary function tests were significantly better. The maintenance of better pulmonary function, coupled with the low mortality, suggested that patients without steatorrhoea have a better prognosis.
At the time the difference was unexplained, but it was eventually shown that the “pancreatic sufficient” patients more often had so-called ‘mild’ gene mutations – a possibility already suggested by Stern who had earlier described seven patients with “Pseudomonas bronchitis”, borderline sweat tests and normal absorption (Stern RC, et al. JAMA 1978; 239:2676-2680). [PubMed]
Dr Gordon Forstner (fig. 16) was the senior paediatrician and gastroenterolgist at the Hospital for Sick Children Toronto, at the time and a major influence on the development of the Cystic Fibrosis service.
M E Hodson, H J Wingfield, J C Batten. Tobramycin and carbenicillin compared with gentamicin and carbenicillin in the treatment of infection with Pseudomonas aeruginosa in adult patients with cystic fibrosis Br J Dis Ches 1983 Jan;77(1):71-7.[PubMed]
Treatment with gentamicin or tobramycin given intravenously with carbenicillin is compared in the treatment of bronchopulmonary infection in older patients with cystic fibrosis. All patients improved on both regimens and there was no observed difference in the two treatments. Pseudomonas aeruginosa could still be isolated from the sputum 3 months after completion of treatment with both treatments. No toxic or allergic side-effects were noted.
1983 Knight RK. Antibiotic doses for bronchiectasis of cystic fibrosis. Lancet 1983; 2:970-971. [PubMed]
One of many publications on CF and various respiratory topics by Dr Ron Knight (fig. 17) a chest physician who worked at the Brompton Hospital

Fig 17. Ron Knight
and subsequently developed his own unit – The Knight Centre – at Frimley Park Hospital, Surrey. He had a reputation for intensive treatment at a time when intravenous antibiotic treatment was not so widely or frequently used. His personal involvement, intensive treatment and good results were greatly appreciated to the extent that the patients and families raised the funds for the Knight Centre at Frimley Park Hospital. The recommendation in this letter to use much larger doses of antibiotics is characteristic of his approach and appropriate for cystic fibrosis.
1983 Carbens NJB, Gosden G, Brock DJH. Microvillar peptidase activity in amniotic fluid: possible use in prenatal diagnosis of cystic fibrosis. Lancet 1983; i: 329-331. [PubMed]
The activities of two amniotic fluid peptidases were significantly depressed in the second trimester amniotic fluid supernatant in the presence of a fetus affected by cystic fibrosis. Eventually David Brock, of Edinburgh, used monoclonal antibody specific for isoenzyme of alkaline phosphatase (see Brock 1983 below). Eventually both methods were superseded by superior DNA based methods.
1983 Desmond KJ, Schwenk WF, Thomas E, Beaudry PH, Coates AL. Immediate and long term effects of chest physiotherapy in patients with cystic fibrosis. J Pediatr 1983; 103:538-542. [PubMed]

Fig 18. Allan Coates Author’s photo
Spirometric and plethysmographic evaluations were performed before chest physiotherapy (CPT) and at five and 30 minutes post-CPT. The pre-CPT measurements after a three-week period with no CPT were compared with the values while receiving CPT on a regular twice daily basis. The findings showed that although there may be little immediate functional improvement when CPT is received on a regular basis, a three-week period without CPT resulted in a worsening of respiratory function which was reversed with renewal of regular chest physiotherapy.
This is one of the few studies evaluating the efficacy of chest physiotherapy. Previously Pryor et al, (1979 above) had evaluated the forced expiratory technique. Subsequently Reisman et al, (1988 below) performed a trial of physiotherapy in Toronto which also showed the benefit of routine physiotherapy. Considering the huge time commitment of the patients and their families to physiotherapy there were surprisingly few publications evaluating the efficacy of physiotherapy treatment.
1983 Brock DJH. Amniotic fluid alkaline phosphatase isoenzymes in early prenatal diagnosis of cystic fibrosis. Lancet 1983; ii: 941-943. [PubMed]

Fig. 18a David Brock
Antenatal diagnosis in families with a known CF child was possible by assay of the microvillus enzymes at 17-18th week of pregnancy. In pregnancies with a CF fetus there was a profound deficiency of one form of alkaline phosphatase (the phenylalanine-inhibitable form). When phenylalanine and homoarginine were used to define the alkaline phosphatase isoenzymes in stored amniotic fluid, 9 out of 10 cases of CF were identified (Brock DH et al, Hum Genet 1984; 65: 248-25. [PubMed]; Brock DH et al, Hum Genet 1988; 78:271-275. [PubMed]
Later, in 1993, David Brock commented that assay of microvillar enzymes in the second trimester amniotic fluid supernatant had a rational physiological basis and had stood the test of time but eventually it was superseded by superior DNA-based methods.
1983 Wilcken B, Brown AR, Urwin R, Brown DA. Cystic fibrosis screening by dried blood spot trypsin assay: Results in 75,000 newborn infants. J Pediatr 1983; 102:383-387. [PubMed]

Fig. 19 Bridget Wilcken ed.ac.uk/alumni
An early report of neonatal CF screening from New South Wales, Australia where CF was confirmed in 35 of 38 infants with persistently raised immunoreactive trypsin (IRT) levels; only 37% of the infants would have been diagnosed by having a family history of CF or by their having meconium ileus.
There were further publications from Bridget Wilcken providing further evidence for the value of neonatal CF screening (Wilcken et al, Arch Dis Child 1983; 58:863-866; Wilcken & Chalmers. Lancet 1985; ii: 1319-1321 and others).
Professor Bridget Wilcken of Sydney, (figure 19) has been and remains a pioneer and constant advocate of neonatal CF screening since the early Eighties.
Bridget Wilcken (figure 19) and her colleagues were one of the first groups to introduce neonatal CF screening in New South Wales using the IRT method described by Crossley and Elliott in 1979 (above), even though there was still considerable discussion as to the benefits which continued into the next Millennium! Screened children in Australia did well compared to non-screened almost certainly as most had CF Centre care, in contrast to the UK where most were treated at their local hospitals – almost certainly accounting for the lack of benefit from neonatal CF screening in a large study in Wales and West Midlands from 1985 for five years (Chatfield S et al, 1991 below).
After considerable pressure from the Cystic Fibrosis Trust, national neonatal screening for CF was approved by the UK Government in 2001 (against the advice of the UK National Screening Committee!) and eventually introduced throughout the UK by 2007.
1983 Bali A, Stableforth DE, Asquith P. Prolonged small-intestinal transit time in cystic fibrosis. BMJ 1983; 287: 1011-1013. [PubMed]

Fig 20. David Stableforth
A lactulose hydrogen breath test was used to measure small intestinal transit time in patients with CF and controls. Seven of 10 CF individuals had high fasting breath hydrogen levels (25-170ppm; normals <20ppm) that fell to normal after prolonged fasting; this indicates bacterial colonisation of the small bowel and was the first report of this occurring in cystic fibrosis. Seven of the 10 patients had prolonged small intestinal transit times (160-390 minutes) compared with controls (50-150 minutes). Again, the first documentation of the prolonged small bowel transit time in CF which was confirmed in subsequent studies.
Increased fasting breath hydrogen and after glucose (Bond JH, Levitt MD. J Clin Invest 1972; 51:1219-1225. [PubMed]) or lactulose (Rhodes JM et al, Scand J Gastroenterol 1979; 14:333-336. [PubMed]) indicates small bowel bacterial overgrowth. A later-than-normal rise after oral lactulose indicates prolonged small bowel transit time as the non-absorbable sugar is fermented by the bacteria in the large bowel at a later stage.
Although these were interesting first reports of small bowel bacterial colonisation and slow transit in CF, the results did not have a major impact on practical management as interest in most CF centres was focused on the chest infection. Fortunately by 2012 there was increasing interest in these matters – both bacterial contamination and inflammation of the small bowel wall which appears to be a relatively frequent occurrence (Smyth et al, 2000 below; Werlin S et al, 2008 below).
Dr David Stableforth (figure 20) started the Birmingham Adult CF Centre initially to continue the treatment of young people with CF who had been attending Professor Charlotte Anderson’s clinic at the Birmingham Children’s Hospital. As Director of the Birmingham Adult CF Centre he developed a major CF Centre at Heartlands Hospital, Birmingham.
1983 Szaff M, Hoiby N, Flensborg EW. Frequent antibiotic therapy improves survival of cystic fibrosis patients with chronic Pseudomonas aeruginosa infection. Acta Paediatr Scand 1983; 72; 651-657. [PubMed]
This is one of the main reports of improved survival at the Danish CF Centre since starting 3-monthly courses of intravenous anti-Pseudomonal antibiotics for chronically infected patients. During the period 1971-75, 51 chronically infected CF patients were treated with IV antibiotics but only when their condition deteriorated. During the period 1976-80, 58 chronically infected CF patients were treated with regular courses of intravenous antibiotics every three months.
The five year survival of patients from the time of the onset of their chronic P. aeruginosa infection increased from 54% in the first period to 82% in the second period. The authors concluded that intensive “maintenance” chemotherapy against P. aeruginosa improves survival and quality of life of CF patients although permanent eradication of chronic P. aeruginosa was not accomplished.
These results were largely ignored outside Denmark even in Europe. True, this was not a controlled trial and there were problems with the cost of the antibiotics, drug allergies, the patients’ time and quality of life and there were other treatments which could have favourably affected the outcome. It is also particularly relevant that long term nebulised antibiotics, that frequently stabilise the respiratory function of chronically infected patients, were not used at the time of this study. Unfortunately, even in 2012, there is still no totally adequate controlled trial of this form of regular IV antibiotic treatment although many clinicians have increasingly adopted a policy of very early intervention – which in practice almost amounts to 3-monthly treatments for some patients. One study from the UK comparing 3-monthly elective with symptomatic treatment showed no advantage in elective treatment but even the “treat when symptomatic” group received 3 courses of IV antibiotics annually (Elborn et al, Thorax 2000; 55:355-358 below).
Perhaps the frequency of intravenous antibiotic treatments is best left to the judgement of the clinician and the CF team who know the patient – and last, but not least, to the patient’s opinion! Certainly, the availability of effective inhaled antibiotic therapy has influenced the management of the patients with chronic P. aeruginosa infection and reduced the need for intravenous antibiotic treatments in some patients.
Professor Niels Hoiby received the ECFS Award in 2012 in recognition of his quite outstanding contribution to the understanding of Pseudomonas lung infection and translating this knowledge into better outcomes for people with cystic fibrosis. He was also recognised for his contribution as the Founding President of the European Cystic Fibrosis Society and his lifelong commitment to the CF center in Copenhagen.
1983 Kirk JM, Adams A, Westwood A, McCrae WM. Measurement of osmolality and sodium concentration in heated cup sweat collections for investigation of cystic fibrosis. Ann Clin Biochem 1983; 20:369-373. [PubMed]
A study from Edinburgh of a new system (Wescor) for sweat collection and analysis with respect to its suitability for the investigation of children suspected to have cystic fibrosis. The effects of iontophoresis current sweat collection time, sweat storage and analysis were examined, and as a result the technique was modified to allow collection and storage of sufficient sweat for sodium and potassium as well as osmolality assays in 10-20 minutes. The small electrodes and speed of the procedure made it practical for use with small children, with a reproducibility of 13-24% (coefficient of variation for whole procedure (also Carter EP et al. 1984; Webster & Barlow, 1981). Most centres did not employ this system.
1984 Pencharz P, Hill R, Archibald E, Levy L, Newth C. Energy needs and nutritional rehabilitation in undernourished adolescents and young adult patients with cystic fibrosis. J Pediatr Gastroenterol Nutr 1984; 3 (Suppl 1): S147-S153. [PubMed]

Fig 21. Paul Pencharz
The energy needs, nutritional status and body composition of six undernourished adolescents and young adults with CF were shown to be 25-80% higher than in healthy individuals of the same age, sex and size. There was significant wasting of adipose tissue. During the short period of refeeding, body weight, fat and potassium all increased significantly, while fat-free body mass and total body nitrogen did not change.
This was the first report from Toronto documenting increased energy expenditure in people with CF. The increased energy expenditure was related to the severity of the chest infection and the increased requirements were lessened when the chest infection was treated. These facts were confirmed in many subsequent studies (Vaisman et al, J Pediatr 1987; 11:496-500; Buchdahl et al, J Appl Physiol 1988; 64:1810-1816; Bowler et al, Arch Dis Child 1993; 68:754-759 all below).
Professor Paul Pencharz (figure 21) is a gastroenterologist and nutritional expert in the Departments of Nutritional Sciences and Paediatrics in Toronto. He has published widely on the nutritional aspects of CF and many other conditions. This present report was the first to clearly document the increased resting energy expenditure in people with cystic fibrosis.
1984 Schoni M, Kraemer R, Ruedeberg A, Lentze MJ, Mordasini RC, Riesen WF, Klay MP, Rossi E. Long-term cimetidine in children with cystic fibrosis: a randomized double-blind study. Pediatr Res 1984; 18:66-70. [PubMed]

Fig 22. Martin Schoni and Mrs Schoni at Artimino CF meeting in 2006
The group from Berne having previously shown some improvement with acid suppression with cimetidine in a few patients (Helvet Paediatr 1981; 36:359-369.) performed this detailed prospective, randomized double-blind study of 38 children with CF designed to evaluate the effectiveness of cimetidine (600 mg/m2) in improving fat absorption and clinical condition. They concluded that cimetidine does not improve fat absorption and has, therefore, no place and no benefit in the treatment of children with CF.
In Leeds, in contrast, we found that cimetidine seemed to improve absorption (Chalmers DM et al, Acta Paediatr Scand 1985; 74:114-117. [PubMed]) with a significant reduction in faecal fat in 17 patients.
However, the acid resistant microspheres Pancrease and Creon were available in the UK from the early Eighties and increasingly used so, for a time, interest in acid suppression to improve the absorption with the older unprotected enzymes waned. Some years later there was a revival of interest in acid suppression, as a means of reducing the dose of enzymes, following the description of fibrosing colonopathy (Smyth et al, 1994 below), considered to be related to excessively high doses of the high strength enzymes introduced in the early Nineties. Also as oesophageal reflux became increasing recognised as a frequent and significant complication particularly in older patients (Feigelson et al, 1975 above; Scott et al, 1985 below) acid suppression was a more frequently prescribed treatment.
Professor Martin Schoni (figure 22) from Berne has been involved in the care of people with CF and also in research since the Seventies and has published widely on the subject. Both he and Richard Kraemer were colleagues of Professor Rossi in the Berne clinic.
1984 Efthimiou J, Smith MJ, Hodson ME, Batten JC. Fatal pulmonary infection Mycobacteriumfortuitum in cystic fibrosis. Br J Dis Chest 1984; 78:299-302. [PubMed]
An early, probably the first, report of an atypical Mycobacterial infection in a young adult with cystic fibrosis. The organism was resistant to all antibiotics and the patient died. Atypical mycobacteria were to become an increasing problem in people with CF.
In a subsequent paper from Royal Brompton Smith MJ et al (1984 below) made a search to determine the prevalence of atypical mycobacteria in their large population of adult CF patients – presumably as a result of experience with this patient. (Also Boxerbaum B. Isolation of rapidly growing mycobacteria in patients with cystic fibrosis. J Pediatr 1980; 96:689-691).
1984 Smith MJ, Efthimiou J, Hodson ME, Batten JC. Mycobacterial isolations in young adults with cystic fibrosis. Thorax 1984; 39:369-375. [PubMed]
Seven of 223 patients with CF admitted to the Brompton Hospital over a six year period had Mycobacteria in their sputum. The organisms isolated were Mycobacterium tuberculosis in three patients, M. chelonei in one, M. fortuitum in one, and unidentified Mycobacteria in two. The diagnosis was not suspected on clinical grounds in any of these patients; in one patient, however, night sweats were a prominent feature before diagnosis. In four of the patients direct sputum smear examination did not reveal the organism, which was grown subsequently in culture.
These were early days for appreciating the role of atypical Mycobacteria in CF and the organisms were present in the sputa of patients with CF more often than previously recognised. Therefore the authors recommended that sputum examination and culture for Mycobacteria should be performed periodically in these patients.
Subsequently problems with these organisms would become more widely recognised and occur as a problem sometimes after lung transplantation when the patients were on immunosuppressive therapy.
Probably as there were fewer adults at that time and so few CF centres for adults, there were no further reports until one from Dublin in 1990 (Mulherin D, et al. Skin reactivity to atypical mycobacteria in cystic fibrosis. Respir Med 1990; 84:273-276. [PubMed]) where the frequency of positive skin reactions was found to be similar as in a control population. One of 43 patients grew an atypical mycobacterium from the sputum.
1984 Isles A, McLuskey I, Corey M. Pseudomonas cepacia infection in cystic fibrosis: an emerging problem. J Pediatr 1984; 104:206-210. [PubMed]
The prevalence of Pseudomonas cepacia infection in a population of approximately 500 patients with CF in the Toronto CF centre, increased from 10% in 1971 to 18% by 1981. The carriage of P. aeruginosahad remained unchanged at 70% to 80% over the same period. Patients infected with P. cepacia had greater impairment of pulmonary function than those with P. aeruginosa alone.
A syndrome characterized by high fever, severe progressive respiratory failure, leukocytosis, and elevated erythrocyte sedimentation rate (“cepacia syndrome”) had occurred in eight patients over the previous three years, with a 62% fatality rate. Because P. cepacia strains are uniformly resistant to ticarcillin, piperacillin, and aminoglycosides, and because ceftazidime is ineffective despite in vitro activity, treatment of these infections was very difficult. Prevention of acquisition and effective treatment of P. cepacia in patients with cystic fibrosis had become a major problem in the Toronto clinic.
This paper from Toronto describes the devastating effect of the introduction of B. cepacia into a CF clinic population very clearly and heralded a new era in CF care. This organism was to have a profound permanent effect on the treatment and social life of people with CF and their families.
The potential for spread between people with CF and the serious, often fatal, consequences (the so-called “cepacia syndrome”) were not fully appreciated in the UK until the early Nineties when John Govan’s publication documented definite person to person spread between people with CF (Govan et al, 1993 below).
From 1993 there was a general introduction of infection control measures in CF Centres in the UK; also there was an end to the North American CF holiday camps which were shown to be an important source of acquisition of the B. cepacia infection.
In Leeds we had three patients who developed serious B. cepacia infections some months after visiting Canadian CF camps. In 1990 we published the first UK paper on B. cepacia in CF, having identified 11 patients with the infection over 6 years since 1984 but we found no evidence of obvious cross infection between our patients when we first identified the infection (Simmonds EJ, et al. Pseudomonas cepacia: a new pathogen in patients with cystic fibrosis referred to a large centre in the United Kingdom Arch Dis Child 1990; 65:874-877. [PubMed]
1984 Winter RJ, George RJ, Deacock SJ, Shee CD, Geddes DM. Self-administered home intravenous antibiotic therapy in bronchiectasis and adult cystic fibrosis. Lancet 1984; i: 1338-1339. [PubMed]

Fig 23. Duncan Geddes CBE
One of the early UK reports of home intravenous antibiotic treatment in cystic fibrosis. Ten patients, eight with CF, and two with advanced bronchiectasis without CF, while being treated in hospital for exacerbations of Pseudomonas infection, were taught how to continue to give themselves intravenous antibiotics at home. In the eight patients, with two or more infective exacerbations within a 12-month period, there was no difference between home and hospital treatments in clinical improvement or in relapse time.
This was a new treatment arrangement and the potential risks of treatment at home caused some people concern at the time. What about the medico legal risks? However, the giving of IV antibiotics at home was a major advance in patient care and a new step in treatment in the UK. Home IV antibiotics had been used in the USA in a few centres some years previously, one from the early Seventies (Rucker et al, 1974 above; Stern RC. Intravenous treatment: where are we and how we got there? In, Doershuk CF, Ed. Cystic Fibrosis in the 20th Century. AM Publishing Cleveland 2001; 93-111).
Professor Duncan Geddes CBE (figure 23) worked at a number of London hospitals including the Royal Brompton where he was involved in many aspects of respiratory disease but particularly with CF both in clinical care and research being a member of the UK Gene Therapy Gene Therapy Consortium. His clear plenary lectures at both European and North American CF conferences were greatly appreciated. Duncan retired in 2008 and in 2012 was awarded the CBE for services to medicine.
1984 Falk M, Kelstrup M, Anderson JB, Kinoshita T, Falk P, Stovring S, Gothgen I. Improving the ketchup bottle method with positive expiratory pressure, PEP, in cystic fibrosis. Eur J Resp Dis 1984; 65:423-432. [PubMed]

Fig 24. The Positive Expiratory Pressure mask

Fig 25. The PEP mask in use
The first report of the Positive Expiratory Pressure (PEP mask) for physiotherapy from Copenhagen that proved to be a popular and effective method of treatment. The mechanics of this method had been evaluated by one of the authors using parts of excised human lungs (Andersen J B et al. Scand J Respir Dis 1979; 60:260-266. [PubMed]. Falk et al studied the acute effects of four different chest physical therapy regimens using a randomised cross-over design in 14 patients with cystic fibrosis. PEP was well accepted by the patients, who preferred treatment with PEP, and the authors suggested PEP be incorporated in chest physical therapy regimens.
(Tyrell et al, 1986 below, for the first UK study of PEP from Nottingham; also Murray JF. The ketchup-bottle method. [Editorial] N Eng J Med 1979; 300:1155-1157). A trail of PEP from George Davidson’s unit in Vancouver had a significant influence on physiotherapy practice in N. America (McIlwaine PM et al. J Pediatr 1997; 131:506-508. below).
1984 Sturgess JM. Structural and developmental abnormalities of the exocrine pancreas in cystic fibrosis. J Pediatr Gastroenterol Nutr 1984; 3 Suppl 1:S55-66. [PubMed]

Fig 26, Jennifer Sturgess
Early signs of a deficiency in exocrine pancreatic tissue at 32-38 weeks post-conceptional age suggest that there is a lack of normal maturation of pancreatic exocrine tissue that occurs in utero, with a degenerative process supervening after birth. The volumes of the acinar and duct lumen are markedly increased, up to 10 fold normal volume in CF subjects. The diagnosis of CF within the first few months of life is difficult when based on conventional or subjective pancreatic histology.
Professor Jennifer Sturgess (1944-2009) (figure 26), the distinguished paediatric pathologist from Toronto, described a method of assessing the pancreatic tissue providing an objective approach that clearly separates CF from control subjects. In this retrospective survey, 93% of CF infants were discriminated from normals; only 2 of 30 cases (7%) were not clearly differentiated from controls.
Earlier studies had reported some CF infants had normal pancreatic histology (Oppenheimer & Esterly, 1973 above).
1984 Reardon MC, Hammond KB, Accurso FJ, Fisher CD, McCabe ER, Cotton EK, Bowman CM. Nutritional deficits exist before 2 months of age in some infants with cystic fibrosis identified by screening test. J Pediatr 1984; 105:271-274. [PubMed]
Data on the first 20 infants identified in the Colorado neonatal CF screening programme. Although birth weights were normal, by a mean age of 5.5 weeks nine infants had a weight more than 20 percentile points less than their birth weight although their dietary intake was normal. Albumin and pre-albumin levels were low in thirteen. Eight had elevated alkaline phosphatase levels; five had low cholesterol levels and stool trypsin was undetectable in nine, low in four and normal in three.
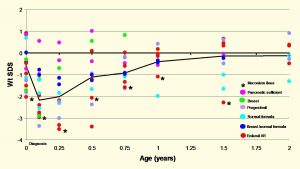
Fig 27. Early weight loss and slow recovery in screened CF infants in Leeds. Wolfe et al. J Cyst Fibros 2003;4(81); S94
The very early onset of nutritional and weight gain problems, even when diagnosed early by neonatal screening, has been observed in a number of series of screened CF infants including our own in Leeds (figure 27). Also the catch up growth period may last for the whole of the first year even with frequent expert dietetic support (Wolfe et al, 2005).
The illegible Right hand side list key for fig 27 from top down – meconium ileus, pancreatic sufficient, breast Pregestemil. Normal formula, Breast/normal formula , Enfamel AR

Fig.27a Keith Hammond
Dr Keith Hammond (Fig. 27a) and his colleagues in Denver Colorado were early into neonatal CF screening and in particular in describing the very early onset of multiple nutritional deficits. The infants detected in their screening programme were studied closely resulting in a number of valuable reports relating to vitamin and nutritional status, pancreatic function, bacteriology and respiratory condition. Keith is one of pioneers of neonatal CF screening.
1984 Wheeler WB, Williams M, Matthews WJ Jr, Colten HR. Progression of cystic fibrosis lung disease as a function of serum immunoglobulin G levels: a 5-year longitudinal study. J Pediatr 1984; 104:695-699. [PubMed]
The children with persistent hypogammaglobulinemia G showed significantly better lung function, better weight for age, fewer hospitalizations for pulmonary exacerbations, less colonization with Pseudomonas aeruginosa, and slower decline in pulmonary functions than did age-matched patients with normal or high IgG levels. Death occurred in five of eight (63%) hypergammaglobulinemia, three of 30 (10%) with normal levels and one of 32 (3%) with low levels. No deaths occurred in the 15 patients with persistent hypogammaglobulinemia.
These data support previous reports that children with cystic fibrosis and hypogammaglobulinemia G have milder lung disease and slower deterioration in pulmonary function than do age-matched patients with normal or elevated immunoglobulin G values. The mechanisms accounting for this finding are unclear (also Matthews et al, 1980 above; Garside et al, 2005; Garside et al, 2007 both below).
1984 Phelan P, Hey E. Cystic fibrosis mortality in England & Wales and in Victoria Australia. Arch Dis Child 1984; 59:71-83. [PubMed]
The contents of this paper had a major influence on CF treatment in the UK.

Fig. 28 Peter Phelan RCH Melbourne
Dr Peter Phelan (1936-) (Fig. 28) is one of the outstanding leaders of paediatric respiratory care and research in Australia and internationally
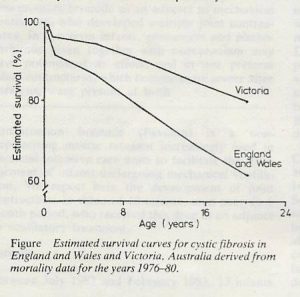
Fig 29. From Phelan &Hey 1984. With permission of BMJ
In Victoria, Australia one death in 125 between the ages of 1 and 14 years was in a child with CF – in England and Wales the was 1 in 44. A newborn child with CF had an 80% chance of surviving to 9 years in England and Wales and to 20 years in Victoria. The better survival in Victoria during the Seventies was attributed to the Australian patients being treated at CF Centres. During the Seventies most UK children with CF were treated at their local hospitals by general paediatricians most of whom saw only a few children with cystic fibrosis.
The authors note “It has been claimed in North America that management in a specialist centre, as in Victoria, is essential for optimal care and recent data from Denmark indicate a much better survival among patients attending a specialist clinic compared with those looked after by their local paediatrician, as happens for most children in England and Wales”.
The data in this report was available before publication through John Dodge who had visited Australia before its publication and the availability of the data was directly responsible for the formation of the British Paediatric Association’s Working Party on Cystic Fibrosis in 1982 whose terms of reference were “to assess the advantages and disadvantages of regional centres for cystic fibrosis”. For summary of the Working Party Report see Jackson 1986 (below).
John Dodge later commented to me that he considers the original data in this report were seriously flawed because the authors compared mean age at death (UK) with standard survival calculations (Australia), but the point they erroneously made was of great value in setting up not only CF centres in the UK but also a CF registry, without which any data are totally unreliable!

Fig. 29 Edmund Hey
Dr Edmund Hey (1934-2009) (figure 29) the co-author of this paper, was a UK consultant paediatrician with previous experience in neonatal physiology and paediatrics who, in 1973, became the first full time consultant respiratory paediatrician at Great Ormond Street Hospital, London; there he met Peter Phelan (figure 28) who spent a six month sabbatical there.
In 1978, on the death of Gerald Neligan (the neonatal paediatrician in Newcastle, UK), Hey was invited to return to Newcastle, as neonatologist which he did.
Later in 1982, Phelan invited Hey back to Melbourne to cover the sabbatical leave of a colleague; it was during this six months that Hey was involved in the present paper. When he returned to Newcastle he concentrated on developing neonatal services and had an outstanding career in neonatology. However, he later said to me (2008) “but I did watch the slow transformation of CF care across the UK with some knowledge that I had probably helped to nudge it into an important change of direction.” This is undoubtedly true!
1985 Wilcken B, Chalmers G. Reduced morbidity with cystic fibrosis detected by neonatal screening. Lancet 1985; ii: 1319-1321. [PubMed]
Cystic fibrosis-related morbidity was evaluated by comparing hospital admissions for CF-related illness in the first two years of life in 40 patients detected by means of neonatal screening and 56 patients born in the three years before screening began. Unscreened patients without meconium ileus had a mean of 27.25 hospital days for CF-related illness, and screened patients a mean of only 3.9 days. There was no trend with time towards fewer days spent in hospital: the change was sudden following the introduction of neonatal screening. The difference was significant and could not be attributed to non-comparability of groups, changes in admission policy, or changes in management. So in Australia neonatal screening significantly reduced CF morbidity in the first two years of life.
The impressive improvement in the outlook for the screened infants, quite obviously related to the introduction of neonatal CF screening, was not accepted by Cochrane reviewers as historical controls were used. However, the advantages of screening were blatantly obvious and acceptable to most experienced CF clinicians who, before neonatal screening, had seen so many CF infants sustain severe irreversible and ultimately fatal lung damage and malnutrition before they were eventually diagnosed and treated.
1985 Castile R, Shwachman H, Travis W, Hadley CA, Warwick W, Missmahl HP. Amyloidosis as a complication of cystic fibrosis. Am J Dis Child 1985; 139:728-732. [PubMed]
Three patients with amyloidosis complicating CF are reported to add to the six patients previously recorded. The presenting problem was proteinuria in five patients, enlarged thyroid in three patients, and hepatosplenomegaly in one patient. The progression of proteinuria to nephrotic syndrome and oedema occurred in eight of the nine patients and indicated a very poor prognosis. The kidneys, adrenal glands, spleen, thyroid gland, liver, heart, and bowel were most frequently involved. Renal involvement was a frequent and devastating complication of amyloidosis in patients with cystic fibrosis.
Amyloidosis is a surprisingly rare complication of cystic fibrosis considering the severity and duration of pulmonary sepsis in most patients.Two cases had been reported by Ristow SC, et al, 1977; 131:886-888 [PubMed]) and others have been reported subsequently. Often renal or thyroid problems are associated.
1985 Levy LD, Durie PR, Pencharz PB, Corey ML. Effects of long-term nutritional rehabilitation on body composition and clinical status in cystic fibrosis. J Pediatr 1985; 107:225-230. [PubMed]
One of a number of papers reporting effects of enteral feeding around this time. Fourteen patients aged 4.9 to 21.5 years with CF who had moderate to severe lung disease, malnutrition, or growth failure were given nocturnal supplemental feeding by gastrostomy tube. Mean follow-up was for 1.1 years (range 0.8 to 2.78 years). Patients were studied to observe the effect of nutritional support on body composition, growth, pulmonary function, and quality of life. A contemporary group of patients with CF was retrospectively pair-matched to the study group. The supplemental feeding resulted in positive changes in body composition and in growth velocity. Weight, as a percentage of standard in the control group, declined by 3% over 1 year, whereas it increased by 2% in the treatment group (p< 0.05). Pulmonary function, assessed as a percent of predicted FVC and FEV1, did not change significantly in the treatment group over 1.1 years, whereas FVC declined by 12% (P<0.01) and FEV1 declined by 13% (P<0.01) in the control group (this is rather alarming decline and far greater than one would expect and is not explained). There was a marked increase in patient ability to participate in activities of daily living, even in those patients in whom pulmonary function deteriorated during the study.
1985 Brock DJH, Befgood D, Barron L, Haward C. Prospective prenatal diagnosis of cystic fibrosis. Lancet 1985;I: 1175-1178. [PubMed]
An immunoassay based on monoclonal antibodies with specificity for the three major isoenzymes of alkaline phosphatase (ALP) was used in second-trimester prenatal diagnosis of cystic fibrosis. When prospective and retrospective data were summed the sensitivity of the test was 91% (39 of 43) and the false-positive rate 6% (5 of 81). The authors concluded that this was probably an acceptable form of prenatal diagnosis of CF for the high-risk mother at the time.
David Brock of Edinburgh pioneered antenatal diagnosis prior to the identification of the probes in close proximity to the CF gene in 1985. He had previously made major contributions to the antenatal recognition of spina bifida.
1985 Littlewood JM, Miller MG, Ghoneim AT, Ramsden CH. Nebulised colomycin for early Pseudomonas colonisation in cystic fibrosis. Lancet 1985;1:865.

Fig 32. Mike Miller (centre) with John Gilbert (our second CF Fellow) and Ann Littlewood (Research Nurse)
Fig 31. Table from the 1985 letter Permission from the Lancet.
The first report of the use of nebulised colomycin (figure 31) to eradicate early Pseudomonas infection and, although only a modest letter, was undoubtedly the most important publication of my career!.
Eradication had previously been thought to be impossible. Subsequently, and according to Niels Hoiby, as a result of this short letter to the Lancet, in Copenhagen they started a controlled trial of early eradication therapy (Valerius et al, 1991 below) and found their results “confirm and extend the preliminary report by Littlewood and colleagues”. We had been using nebulised colomycin in Leeds since about 1983/84 when Pseudomonas was first isolated to attempt eradication and after treating seven patients it was quite apparent that in some cases the organism was cleared from the sputum contrary to current teaching; so we reported our anecdotal experience in this letter to the Lancet.
Dr Michael Miller (figure 32) was one of the first CF Research Fellows in the UK funded by the Cystic Fibrosis Trust. He was with us in Leeds for over 2 years and, with the CF Nurse Specialist Teresa Robinson, played a very important part in establishing our paediatric CF Unit at St James University Hospital. Mike subsequently was consultant paediatrician in Hudderfield, Yorkshire and eventually consultant in Paedaitric Palliative Medicine at Martin House Hospice in Boston Spa, Wetherby, Yorkshire.
The slow introduction of early eradication therapy, particularly in North America, is difficult to understand although perhaps one reason was that, in the early Eighties, the serious long term consequences of chronic Pseudomonas infection were only just being clearly described; most publications documenting the unfavorable outlook for patients with chronic Pseudomonas infection did not appear until the early Nineties (Kerem E et al, 1990. [PubMed]; Henry et al, 1992 [PubMed]; Hudson et al, 1993 [PubMed]; Pamucku et al, 1995. [PubMed]; Frederiksen et al, 1996. [PubMed]; Kosorok et al, 2001. [PubMed]).
However, it is surprising that in a Cochrane Systematic Review of early Pseudomonas eradication treatment as late as 2006, the reviewers, while noting that the organism was eradicated in some patients, were still reluctant to admit that eradicating the organism had a favorable long term effect – presumably ignoring a great deal of published work attesting to the adverse effects of chronic P. aeruginosa infection mentioned above. Some clinicians, including this paediatrician, have mixed feelings about the Cochrane Sytematic Reviews!
1985 (1990) Yacoub MH, Banner NR, Khaghani A, FitzGerald M, Madden B, Tsang V, Hodson M. Heart-lung transplantation for cystic fibrosis and subsequent domino heart transplantation. J Heart Transplant 1990; 9:459-467. [PubMed]

Fig 32a. Magdi Yacoub
A major advance, for those who had reached the end stages of their disease (an FEV1 predicted of less than 30%), was the successful introduction of heart-lung transplantation in 1985, pioneered by Sir Magdi Yacoub (1936- ) (figure 32a) and his team in London (Yacoub et al, 1990; Scott et al, 1988 below) and Mr John Wallwork and his team in Cambridge. The possibility of successful treatment in what were previously the terminal stages of the condition had a major influence on both prognosis and the treatment of severely affected individuals.
The first results of heart-lung transplantations were quite remarkable and were related both to surgical skills, concentrated medical expertise in assessment and after care and also to more successful immunosuppressive therapy to prevent rejection of the transplanted organs. Later double lung transplants became more popular (Pasque et al, 1990 below) and are now the favoured operation.
Dr Norman Shumway laid the groundwork for heart lung transplantation with his experiments into heart transplantation at Stanford in the mid 1960s. Shumway conducted the first adult heart transplant in the US in 1968. Building on his research at Stanford, Dr Bruce Reitz performed the first successful heart–lung transplant at Stanford in 1981 on Mary Gohlke a lady with chronic pulmonary hypertension. The transplant team at Stanford is the longest continuously active team performing these transplants.
(please see also 1985 “Megapapers ” section of this website for more images and text on the history of transplantations)
Living donor lung transplants have proved successful in some centres. On living donor lung transplants Professor Hodson wrote that Professor Starnes from Los Angeles carried out the first successful transplant using living lobe donors in 1990. Initially this option was chosen as a last resort for young people about to die on the transplant waiting list and the results were not encouraging. However as selection of cases, preparation for surgery and post-operative care improved, so did survival of the cases treated with LL transplant. In 1996 Prof Starnes reported a 75% one-year survival rate for 20 patients with CF. This survival was similar to that of patients receiving a heart/lung or a bilateral sequential lung transplant (CL). He reported no mortality among the donors. The great advantage of using this technique is that the CF patient has a chance to be transplanted instead of a 50% chance of dying on the waiting list. (see also Cohen & Starnes, 2001 below; Barr ML, Schenkel FA, Bowdish ME, Starnes VA. Living lobar lung transplantation: current status and future directions. Transplant Proc 2005; 37:3983-3986.
1985 Auerbach HS, Williams M, Kirkpatrick JA, Colten HR. Alternate-day prednisone reduces morbidity and improves pulmonary function in cystic fibrosis. Lancet 1985; 2:686-688.[PubMed]
Children aged one to 12 years with CF received 1-2 mg/kg of prednisone or placebo on alternate days for four years. The prednisone group had better lung function, lower ESR and IgG levels and required only nine admissions compared with 35 in the control group. There were no steroid induced side effects noted in this initial report (but these appeared later).
Later this and another similar study (Eigen et al, J Pediatr 1995; 126:515) both reported significant side effects including impaired glucose metabolism, cataracts and impaired growth – the latter being permanent in boys aged 18 years and older whose mean height was 4 cm less in the prednisolone treated patients (Lai HC et al. N Engl J Med 2000; 342:851-859. [PubMed] (Also re. steroid treatment see Pantin CFA et al. Thorax 1986; 41:34-38 [PubMed] and Greally P et al. Arch Dis Child 1994; 71:35-39 [PubMed] for effects of a course of oral prednisolone in adults with CF).
This paper excited a great deal of interest at the time but later this use of steroids, even on an alternate day basis, was shown to have far too many side effects (including osteoporosis and diabetes) to be used even though alternate day prednisolone was considered to have fewer side effects than daily dosing.
1985 Conway SP, Miller MG, Ramsden C, Littlewood JM. Intensive treatment of Pseudomonas chest infection in cystic fibrosis: a comparison of tobramycin and ticarcillin, and netilmicin and ticarcillin. Acta Paediatr Scand 1985; 74:107-113. [PubMed]

Fig 33. Steven Conway
Seventeen cystic fibrosis patients aged 3.1 years to 19.8 years had 30 courses of intensive intravenous antibiotic treatment for exacerbations of their chronic Pseudomonas chest infection. The combination of netilmicin and ticarcillin was compared with tobramycin and ticarcillin in an open study. A significant subjective and objective improvement occurred in all patients. Pseudomonas was cleared temporarily from the sputum in 11 out of the 30 courses of treatment (37%) but, as expected, soon returned in all. There was no significant difference between the netilmicin and tobramycin groups, nor evidence of sustained renal or ototoxicity. Intensive therapy of Pseudomonas chest infection in cystic fibrosis patients is described in detail.
The main purpose of our publishing this paper, in addition to the trial of netilmicin, was to document the fine details of a course of intravenous antibiotic therapy for those UK paediatricians who, like ourselves, were learning all the time how to best treat children with cystic fibrosis. At this time in 1985 many children with CF in the UK were still cared for at their local hospitals by general paediatricians and had no contact with a CF centre. We hoped this detailed account of treatment would be of help in treating their CF patients.

Fig.33a Steve and Ella Conway with Jim & Ann Littlewood in 2018
Dr Steve Conway’s (figure 33, 33a). This is one of his early papers on cystic fibrosis when he was working with me at St James’s in Leeds. We were very fortunate that Steve remained in Leeds and eventually became Lead Clinician in CF of both the Paediatric and Adult CF units in Leeds. He established the Adult CF unit initially at Seacroft Hospital Leeds and when I retired from the Paediatric Unit 1n 1997 he also took over the Paediatric Unit at St James’s. Steve and his colleagues made major clinical and research contributions to CF care. Also a graduate in English Steve was a prolific and skilled writer and researcher also making many contributions to national and international CF organisations. He was/is a good friend and colleague and was a major force in the development of the Leeds CF services; with Steve and his wife Ella my wife and I enjoyed many times over many years. Steve retired from the NHS in 2014.
1986 Pantin CF, Stead RJ, Hodson ME, Batten JC. Prednisolone in the treatment of airflow obstruction in adults with cystic fibrosis. Thorax 1986; 41:34-38. [PubMed]

Fig 34. Charles Pantin. Author’s photo
Oral prednisolone 0.48 mg/kg body weight per day for 3 weeks caused no significant improvement in lung function. However, atopic subjects showed an improvement in evening recordings of peak expiratory flow rate (PEF) while taking prednisolone (p <0.05). Significant deterioration in forced expiratory volume in one second (FEV1) and forced vital capacity (FVC) were seen after withdrawal of the prednisolone. Two patients developed pneumothoraces while taking prednisolone.
Dr Charles Pantin (figure 34) General and Respiratory Physician, worked first in the adult CF unit at the Brompton Hospital in London and subsequently developed the North West Midlands Cystic Fibrosis Centre. He has also a PhD in applied mathematics and has published extensively on respiratory function and occupational asthma. Charles retired from the NHS in 2011 and became Chairman of the Penkhull Residents Association .
1986 Littlewood JM. An overview of the management of cystic fibrosis. J R Soc Med 1986; 79 (Suppl. 12): 55-63. [PubMed]
A detailed review of treatment in 1985 at the first of many annual meetings on CF organised over more than 25 years, at the Royal Society of Medicine, London by Prof. Tim David of Manchester. Although by now an experienced general paediatrician, I was a relative newcomer to CF compared with some of the audience. However, I had seen many new patients for full assessments over the previous five years from many different hospitals – well over a 100 new patients by 1984. Drawing from this experience and the literature, I tried to describe the areas where treatment appeared to make a significant difference to the long term outlook.
A major problem was to communicate the established facts to the hundreds of clinicians each of whom was responsible for the long term management of only a few CF patients, and each of whom had numerous conflicting demands on his/her time ranging from neonatal intensive care to child abuse. Such paediatricians rarely had the opportunity to attend major CF meetings in Europe or North America as they had so few affected patients. So this was a very detailed account of modern CF care including a plea for some form of CF centre specialist care for all – either full care or shared with the local hospital as already recommended by the British Paediatric Association Working Party. There was still considerable opposition to the concept of CF centre care by many UK paediatricians.
I concluded at the time that “if the outlook for CF patients in the UK was similar to that expected in large CF centres both here and overseas, these suggestions (of some CF centre care for all) would be superfluous. However, it is not and until such time as it is we must give such an arrangement a trial for the sake of CF patients and their families”.
I felt very strongly about this issue having by this time seen so many children with CF referred to Leeds for Comprehensive Assessment during the preceding 5 years who had been severely under-treated by modern standards (Littlewood et al, Comprehensive clinical and laboratory assessment in cystic fibrosis. In: Lawson D, editor. Cystic Fibrosis: Horizons. Chichester: John Wiley, 1984:266; Littlewood et al, 1988 below; Littlewood 1993 below).
1986 Jackson ADM. Working Party on Cystic Fibrosis. Arch Dis Child 1986; 61:724.

Fog. 35 A working dinner.(L. to R.) Tony Jackson (talking), Jim Littlewood (listening), Sir John Batten (checking his watch) and David Lawson (smoking a cigarette).
Dr Tony Jackson, (1918-2005) (figure 35) who prepared this summary, was a paediatrician in London and involved with CF for many years working at the Queen Elizabeth Hospital Hackney and the London Hospital. He qualified at the Middlesex in 1943 and after war service in the RAMC trained in paediatrics, being appointed first to Epping Hospital and later to Queen Elizabeth Hospital Hackney, Great Ormond Street and the London Hospital. He was the longest serving officer of the British Paediatric Association and between 1967 and 1983 was secretary, assistant secretary or treasurer. He was involved with many national and international paediatric committees and latterly was Chairman of the UK Cystic Fibrosis Trust’s Research and Medical Advisory Committee until 1994 when he persuaded me to take over from him.
The present report of the Working Party on Cystic Fibrosis notes the difference in survival between centres the best being from Melbourne with an 80% survival to 20 years, compared with England and Wales where 80% survived to only 9 years. 99% of UK paediatricians responded to a national survey with details of 4557 patients. Only 16 centres treated more than 50 patients and only 1800 patients (46.5%) were on the records of these 16 centres. The working party supported the principle of specialised centres for CF which had been accepted both by the WHO and the International Cystic Fbrosis Association, but suggested that shared care would be appropriate for some children depending on local arrangements. Minimum staffing levels were suggested and close cooperation with adult physicians was recommended as was collaboration between CF Centres in data collection, clinical trials and research.
The data on the better survival in Victoria, Australia than in England and Wales was a major factor in the formation of this UK Working party (Prof John Dodge commented on this data – “the original data were seriously flawed because they compared mean age at death (UK) with standard survival calculations (Australia), but the point they erroneously made was of great value in setting up not only CF centres in the UK but also a CF registry, without which any data are totally unreliable!
Unfortunately, the council of the British Paediatric Association initially rejected the report, the members of the council, we believe misguidedly, maintaining that children with CF could be quite adequately cared for by a general paediatrician at their local hospital. Nonetheless, this was a highly influential report which was published and eventually had a major effect on the development of CF centres throughout the UK, thus steadily improving the care and survival of children with CF.
The CF Survey, which the Working Party initiated, produced useful data on numbers and survival until replaced in 1995 by the UK CF Database in Dundee and then eventually by the UK CF Trust’s CF Registry in 2007- the latter using the North American Port CF system. (Also Dodge et al and BPA Working Party on Cystic Fibrosis. BMJ 1988; 297:1599-1602 for full report and details of a follow-up survey below).
1986 David TJ. Potential practical and legal problems with home administration of intravenous antibiotics for children with cystic fibrosis. In: David TJ, ed. Cystic fibrosis in children. Practical and legal aspects of intravenous antibiotic administration in the home. Amsterdam: Excerpta Medica, 1986:3-14.

Fig 36. Tim David
Report of a meeting organised by Professor Tim David of Manchester (figure 36) to discuss the practical and legal details of home intravenous antibiotic treatment which was increasingly used by CF centres in the UK for the giving treatment at home by parents or patients. The legal situation surrounding such treatment was not entirely clear at the time.
(see also Rucker et al, 1974 above; Winter et al, 1984 above; Gilbert et al, 1988 below; Stern RC. Intravenous treatment: where are we and how we got there? In, Doershuk CF. (Ed.) Cystic Fibrosis in the 20th Century. AM Publishing Cleveland 2001; 93-111).
1986 Kelleher J, Goode HF, Field HP, Walker BE, Miller MG, Littlewood JM. Essential element nutritional status in cystic fibrosis. Human Nutr: Applied Nutrition 1986; 40A: 79-84.[PubMed]

Fig 37. Jerry Kelleher
The late Dr Jerry Kelleher (figure 37) was Senior Lecturer in Biochemistry in Professor Losowsky’s Department of Medicine at St James University Hospital in Leeds. He, with Mr Mike Walters and other members of that department, made a major contribution to the gastrointestinal and nutritional research from the Regional Paediatric CF unit at the hospital from our first research paper and many subsequent studies with the Department of Medicine at St James’s.
In this present study calcium, magnesium, iron, copper and zinc were measured in 117 patients with CF of wide age range. All the minerals were normal except the iron which was frequently low. There was no evidence that the degree of malabsorption, clinical grading or degree of pulmonary involvement influenced the essential element status as assessed by serum levels.
A later paper recording the iron status of 165 patients with CF attending Leeds for Comprehensive Assessment confirmed that 41 (32%) had low serum ferritin levels although the iron status did not correlated with the clinical features (Ehrhardt E et al. Fig 37Arch Dis Child 1987; 62:185-187). A number of subsequent publications dealt with iron status.
1986 Farrell M, Law HY, Rodeck CH, Warren R, Stanier P, Super M, Lissens W, Scambler P, Watson E, Wainwright B, et al. First-trimester prenatal diagnosis of cystic fibrosis with linked DNA probes. Lancet 1986; i: 1402-1405. [PubMed]
More accurate antenatal diagnosis using the new linked probes was described in 1985. Linkage analysis with cloned gene probes has shown that the mutation causing cystic fibrosis was located in the middle of the long arm of chromosome 7. In this paper first-trimester diagnosis of cystic fibrosis is reported in four informative families and second-trimester diagnosis in one family with fetal DNA prepared from chorionic villi, hybridised with the tightly linked DNA probes, pJ3.11 and met. Risk calculations show that the expected false-negative and false-positive rates are approximately 2% and 6%, respectively, for typical nuclear families with one affected living child.
The authors considered existing probes to be sufficiently informative now to allow full diagnosis in about two-thirds of couples presenting with at least one affected child. In half of the remainder, the inheritance of one parental mutant chromosome could be deduced.
1986 Pedersen SS, Koch C, Hoiby N, Rosendal K. An epidemic spread of multiresistant Pseudomonas aeruginosa in a cystic fibrosis centre. J Antimicrob Chemother 1986; 17:505-516.[PubMed]
Early in 1983 an epidemic of a Pseudomonas aeruginosa resistant to aminoglycosides, carbenicillin, ureidopenicillins, ceftazidime, cefsulodin and imipenem occurred in the Danish CF centre. Most of the epidemic could be attributed to a specific nosocomial strain by means of O-grouping and phage- typing. This strain was present in the centre at a low frequency in 1973 and developed resistance during repeated courses of chemotherapy. The epidemic was stopped by isolating the patients with the resistant strains. Restrictive and selective use of antibiotics was not sufficient to eradicate the resistant strains, which persisted in 42% of the patients. The extensive use of the third generation cephalosporins in the clinic was probably responsible for inducing and selecting for the resistant strains. Clustering of patients in the centre facilitated the spread. First-line use of older beta-lactam antibiotics, close bacteriological monitoring and prompt isolation of patients with resistant strains was implemented.
The Danish CF centre was unusual in giving regular 3-monthly courses of IV antibiotics to all their patients who were chronically infected with Pseudomonas aeruginosa (Pedersen et al, 1987 below). It is interesting that in the Liverpool paediatric CF clinic the routine use of ceftazidime monotherapy was associated with the development of a resistant Pseudomonas aeruginosa with epidemic spread amongst the patients there (Cheng et al, 1996 below).
1986 Brett MM, Ghoneim ATM, Littlewood JM, Losowsky MS. Development of enzyme linked immunosorbent assay (ELISA) to detect antibodies to Pseudomonas aeruginosa cell surface antigens in sera of patients with cystic fibrosis. J Clin Path 1986; 39:1124-1129. [PubMed]

Fig 39. Controls were non-CF children. Gp 1. never had Pseudomonas. Gp 2. Intermittent Pseudomonas. Gp 3 Chronic Pseudomonas infection. With permission BMJ.

Fig. 38 Moira Brett
Dr Moira Brett (figure 38) was at the time research immunologist at the Leeds CF unit. The 7 most common strains of P. aeruginosa from our patients were used as antigens in the ELISA test that she developed. The test was specific for Pseudomonas aeruginosa with no cross reaction with other gram negative organisms. We suggested the test may be useful in monitoring progress of P. aeruginosainfection in CF patients; and so it proved to be as subsequent publications confirmed (Brett et al, 1986 below; Brett et al, 1987; Brett et al, 1988; Brett et al, 1988; Brett et al, 1990; Brett et al, 1992). The test has been in use in the Leeds clinic since 1986 until recently replaced by a commercial kit.
1986 Brett MM, Ghoneim ATM, Littlewood JM. Serum antibodies to Pseudomonas aeruginosa in cystic fibrosis. Arch Dis Child 1986; 61:1114-1120. [PubMed]
Serum IgG antibodies to Pseudomonas aeruginosa cell surface antigens were determined by enzyme linked immunosorbent assay (Brett et al, 1986 above). Titres in patients without CF were low (140-235). Those in patients with cystic fibrosis who were chronically infected by P. aeruginosa were very high (1100-20,500), while patients who grew the organism intermittently had lower titres (160-4400) (figure 39). Longitudinal studies showed that raised titres were observed at a very early stage of infection. High titres were associated with a poor clinical state, while low titres were associated with a better clinical state in both chronic and intermittently infected patients with cystic fibrosis. These results suggested that this test is a specific and sensitive measure of the severity and progress of the different stages of pulmonary infection by P. aeruginosa in patients with cystic fibrosis
Dr Moira Brett (figure 38) at this time was a research immunologist who worked in Leeds with the CF unit at St James University Hospital from the mid-Eighties until the early Nineties. She developed the ELISA test for Pseudomonas antibodies and published a number of papers on the use of the test (Brett et al, 1986; Brett et al, 1987; Brett et al, 1988; Brett et al, 1988; Brett et al, 1990; Brett et al, 1992). We found the test a valuable aid to clinical management and it has been used continuously in the Leeds CF unit since the first report in 1986.
It is surprising that many CF Centres still do not routinely estimate Pseudomonas antibodies in their patients with cystic fibrosis. It is a very useful test. We came to the view that the persistent absence of antibodies is very strong evidence that the patient does not have an unrecognised pulmonary Pseudomonas infection; a high level confirms that chronic infection is present; a rising level suggests that the intensity of treatment should be increased; a progressively higher level indicates a poor prognosis; an increased level in a patient who has negative respiratory cultures suggests there is Pseudomonas infection present and is an indication for bronchoscopy to identify the organism. Repeatedly negative monthly respiratory cultures (cough swabs or throat swabs) and negative Pseudomonas antibodies are probably a better indication that Pseudomonas is absent from the airways than a “one off” bronchoalveolar lavage.
1986 O’Halloran SM, Gilbert J, McKendrick OM, Carty HML, Heaf DP. Gastrografin in acute meconium ileus equivalent. Arch Dis Child 1986; 61:128-130. [PubMed]
Gastrografin is a radiological contrast medium containing sodium diatrizoate, meglutamine diatrizoate and iodine plus flavouring and wetting agents. It is hypertonic with minimal absorption from the intestine.
This is the first description from the Liverpool CF unit of the use of oral gastrografin as an effective and subsequently widely used treatment for distal intestinal obstruction syndrome, which, for some reason, was particularly common in the Liverpool CF clinic affecting 37% of the children. In one year 37 of 40 episodes were treated with a single oral dose of gastrografin with an 81% success rate.
It is interesting, and quite unexplained, that fibrosing colonopathy was later described in Liverpool children (Smyth et al, 1993 below) where it was more common than in any other UK CF centre.
1986 MacLusky I, Levison H, Gold R, McLaughlin FJ. Inhaled antibiotics in cystic fibrosis: Is there a therapeutic effect? J Pediatr 1986; 108:861-865. [PubMed]
The Toronto CF centre, under the leadership of Henry Levison, critically examined a number of the treatments used at the time confirming the value of some (physiotherapy – Reisman JJ 1988 below) and failing to confirm the benefit of others (nocturnal mist tents – Chang N et al. 1973 above).
This present study reviewed the evidence for the use of aerosol antibiotics and concluded that many studies were unsatisfactory and the results contradictory and that until additional well-controlled trials were completed their routine use was not justified because of cost, potential side effects and the propensity to select resistant organisms.
By this time in in 1986 in the UK there was increasing use of inhaled antibiotics following the very convincing 1981 paper on nebulised gentamicin and carbenicillin which had a quite obvious significant beneficial stabilising effect in some patients chronically infected with Pseudomonas (Hodson et al, 1981. above). Also, the preliminary observation from Leeds that inhaled colomycin would eradicate early Pseudomonas infection (Littlewood et al, 1985 above) had already been published but was ignored by MacLusky et al.
As it turned out, both the routine use of anti-Pseudomonas antibiotics to suppress chronic infection as recommended by Margaret Hodson in 1991 was 18 years later eventually shown to be effective using tobramycin formulated for inhalation (TOBI) in an acceptable trial (Ramsey BW et al, 1999. below) as was the early use of colomycin to eradicate early infection (Littlewood et al, 1985; Valerius et al, 1991). Both proved to be effective, valuable, proven treatments and eventually widely accepted and used for people with CF in many countries.
1986 Monorails EL, Korpela R. Double blind evaluation of lignocaine-prilocaine (EMLA) in children. Effect on the pain associated with venous cannulation. Brit J Anaesth 1986; 58:1242-1245. [PubMed]
Fig 41. Application of EMLA
Fig 40. Application of EMLA
The application of EMLA (Eutectic Mixture for Local Anaesthetic) local anaesthetic cream (lignocaine-prilocaine cream) to the skin and its occlusion for an hour before venepuncture (figures 40 & 41) was a major advance and was subsequently widely used permitting virtually painless venepuncture in the majority of children. This report was of a trial in 60 children, who incidently did not have CF, using EMLA cream on the skin prior to venous cannulation before surgery; EMLA was clearly superior to placebo cream in preventing the pain of venepuncture.
The introduction of EMLA cream was a very welcome major advance both for the children with CF, who required frequent venepuncture for blood specimens and also for insertion of IV lines for antibiotic treatment; also it was welcomed by the doctors regularly inserting the IV needles – at times a very stressful business for both child, parents and the doctor!! Some children would even come to our clinic with EMLA cream already applied to both arms “just in case” someone decided on a blood test!! (Also Hallen B & Upfield A. Anaesthesiology 1982; 57:340; Clarke S & Radford M. Arch Dis Child 1986; 61:1132-1134; Halperin DL et al. Pediatrics 1989; 84:281-284 all supporting the effect of EMLA cream).
Later nitrous oxide inhalations (Entonox) were used in some units for children before venepuncture (Mills HL et al. 2001 below; Kanagasundaram SA et al, 2001 below; Williams V et al. 2006 below). These measures developed in view of the new need for life long repeated venepuncture in children with CF. So EMLA was undoubtedly one of the major advances in patient care of the decade!!
1986 Orenstein DM, Wasserman AL. Munchausen syndrome by proxy simulating cystic fibrosis. Pediatrics 1986; 78:621-624. [PubMed]

Fig. 41a David Orenstein CF Foundation
Professor Sir Roy Meadow described Munchausen by proxy in 1977 (Meadow R. Munchausen syndrome by proxy. The hinterland of child abuse. Lancet 1977; ii: 343-345. & Meadow R. Munchausen syndrome by proxy. Arch Dis Child 1982; 57:92-98).
This is the first report where CF was the falsified disorder. The mother falsified the history of her child, cunningly altered sweat tests and stool fat analyses and stole sputum from patients with CF to make her child appear to have cystic fibrosis. Previous reports of the syndrome had concerned single signs or symptoms but the present case involved the complex features of a multisystem genetic disorder.
Subsequently nine further reports of Munchausen or Munchausen by proxy with CF of the falsified disorder appeared in the literature up to 2005.
David Orenstein (Fig. 41a) is Professor of Pediatrics, University of Pittsburg School of Medicine. He is the Antonio J. and Janet Palumbo Professor of Cystic Fibrosis, and CF Center Director Emeritus at Children’s Hospital of Pittsburgh. A CF doctor since 1975, he has published more than 200 articles and chapters. Of his three CF-related books, Cystic Fibrosis: A Guide for Patient and Family is in its 4th edition, and is used in many CF centers.
1986 McDowell HP, Hart CA, Martin J. Implantable subcutaneous venous catheters. Arch Dis Child 1986; 61:1037-1038. [PubMed]
The first report of the use of the Port-A-Cath totally implantable venous access device in 12 child oncology patients from Dr John Martin’s paediatric oncology unit in Liverpool; there were fewer complications than with the other central venous catheters and long lines currently in use.
At this time increasing numbers of children with CF were having repeated courses of two weeks intravenous antibiotics; problems with venous access were an increasing and major problem.

Fig. 41b John Martin
This report from Liverpool prompted us in Leeds to combine with our paediatric oncology colleagues at St James University Hospital, to use the Port-A-Cath both in children with CF and those with oncology problems (Essex-Cater et al, 1989 below). Eventually Port-A-Caths and other implantable venous access devices became widely used in all CF Centres for both children and adults requiring regular course of intravenous antibiotics. They were undoubtedly a major advance in treatment.
John Martin (1935-2020) (Fig.41b) was consultant paediatric oncologist at Alder Hey Children’s Hospital, Liverpool where he established a pioneering unit, providing both excellent clinical care and support for children and their families by using specialist nurses and social workers who helped establish paediatric palliative care. It seems appropriate he was the first to Port-A-Caths in children.
1986 Ferguson A, Merrett TG, Littlewood JM, Bolderson I. IgE antibodies to foods are not a feature of cystic fibrosis. Human Nutrition – Clinical Nutrition 1986; 40:255-258. [PubMed]

Fig. 41 Anne Ferguson history.rcplondon.ac.uk
It had been suggested that patients with cystic fibrosis have abnormal immune responses to foods. In collaboration with Dr Anne Ferguson of Edinburgh, we (Ian Bolderson) measured IgE antibodies to inhalants and foods (by RAST) in 105 patients with cystic fibrosis aged between eight months and 28 years. Serum IgE was elevated (greater than 180 kU/l) in 21 (20%) patients. In 43, (41%) IgE antibodies were detected in serum. The majority of positive results were with house-dust mite, grass pollen or Aspergillus. Only four of the patients had a positive RAST to a food – one to milk, one to wheat and two to egg. On the basis of high cserum IgE or positive RAST results, 44.8 per cent of the patients were atopic and the frequency of atopy was age-related, being higher in patients aged four years or more. However, the presence of food antibodies was unrelated to age.
This study confirms the high prevalence of atopy in patients with cystic fibrosis but unequivocally demonstrates that the presence of IgE antibodies to foods in their serum is rare. Of course, we had previously observed that many children with quite obvious clinical food intolerance (proved by withdrawal and challenge) have neither positive skin tests nor raised IgE levels nor positive RAST to foods (Minford AMB et al. Food intolerance and food allergy in children: a review of 68 cases. Arch Dis Child 1982; 57:742-747 [PubMed]).
Professor Anne Ferguson (1941-1998) (Fig. 41c) was one of the UKs most distinguished and respected gastroenterologists.
1986 Tyrrell JC, Hiller EJ, Martin J. Face mask physiotherapy in cystic fibrosis. Arch Dis Child 1986; 61:598-611. [PubMed]

Fig 43. Joan Hiller

Fig 42. Jenny Tyrrell
This study was coordinated by the late Dr Jenny Tyrell (1952-2012) (figure 42) then the CF Research Fellow in Nottingham. Later Jenny was paedaitric consultant in the city of Bath, UK where she started a CF clinic and had a distinguished career.
This study evaluated the use of the ‘PEP’ mask with forced expiratory coughing which was compared with conventional physiotherapy over a one month period. No difference was shown in symptom scores, sputum production, or simple lung function tests. The mask was well accepted and allowed independent treatment by older patients.
The PEP mask became a popular method physiotherapy giving some degree of independence to the patient (also Falk et al, 1984 above and many subsequent papers supporting the use of the PEP).
This paper was from Dr Joan Hiller’s Paediatric CF Unit at Nottingham City Hospital. Dr Joan Hiller (figure 43) founded the Nottingham Paediatric CF Unit having previously worked in the USA with Dr Nancy Huang in Philadelphia.
1986 Pitts-Tucker TJ, Miller MG, Littlewood JM. Finger clubbing in cystic fibrosis. Arch Dis Child 1986; 61:576-579. [PubMed]
We were interested in physical signs which may be useful and were impressed by the study of Sinniah & Omar, using the shadowgram, outlining the finger as a silouette on a screen using an overhead projector, to quantitate finger clubbing (1979 above).

Fig 43. Jeanette Firth
Jeanette Firth (figure 43) the Leeds CF Unit Nurse measured finger clubbing in 73 of 105 patients with CF undergoing full Comprehensive Assessment at the Leeds Regional Paediatric CF Unit. Predictably, the degree of clubbing correlated well with the chest X-ray score, indices of pulmonary function and infection but not with weight, height, age, liver function, or degree of fat malabsorption. The presence of clubbing in a person with CF suggests appreciable pulmonary involvement. Most probably progression indicates deterioration in pulmonary state. In both instances increased efforts should be made to treat the infection.
This study was first presented by our CF Research Fellow Dr Mike Miller at the European CF Working Group Meeting in Israel. There was much hilarity that the doctors in Leeds had just realised clubbing was associated with chest problems!!! However, despite the hilarity it was one of the most discussed presentations of the session. People seemed hungry for some clinical signs that would be helpful in their work. Others later observed that finger clubbing regressed in some patients after successful lung transplantation.
1987 Levison H, Garner D, MacMillan H, Cowen L. Living with cystic fibrosis: patient, family, and physician realities. Compr Ther. 1987; 13:38-45. 3315409 [PubMed]

Fig 44. Henry Levison
The challenge for patient, family, and healthcare professionals alike is to separate the disease’s insidiousness from the potential the patient and family have to enjoy life within the patient’s abbreviated life span. We must emphasize that most patients with CF and their families do make a successful psychological adjustment. Simultaneously, parents must fulfill the varied physical and psychological needs of the child. A young adult patient with CF summarizes the patient’s task: “Projecting a life goal, living it, having the goal altered by luck and by fate, accomplishing that goal, and then reflecting on what has been done. That is life. That is a life compressed for us; CF is myopic. We cannot look through the long vista of life. That is disappointing, frustrating, and cursed at. We can live now. We can do the best we can. Set goals that fit on our playing field and accomplish them.” An emotionally adjusted family makes the patient’s task possible. Achieving the goal of adequate adjustment also depends on the physician’s having the medical expertise to manage this complex multisystem illness and the ability to make the medical knowledge comprehensible for patient and family.
An interesting article from Dr Henry Levison (figure 44) Chair of the Respiratory Division at the Toronto Children’s Hospital from 1985-94. He lead the NIH funded asthma research programme (Childhood Asthma Management Programme) from 1991-97 and with many distinghuished colleagues at the hospital produced some 100 publications on various aspects of CF and over 300 in total. Undoubtedly one of the most active clinical and research paediatricians in N. America. In 2011 Henry was awarded the third annual Victor Chernick Award recognizing life time achievement in the field of pediatric respirology “an icon in this field in Canada. Dr. Levison lead a distinguished career in respiratory medicine at The Hospital for Sick Children, where he provided training and guidance to many a physician!”
In 1980, at the time of the North American CF Conference in Toronto, Dr Archie Norman, his wife and myself visited Henry in his office/laboratory which contained a vast amount of scientific apparatus amongst which he saw a child with asthma. I made the mistake of asking if he checked the CF children for allergies. “Oh no – you’re not a b….y allergist?” Although I denied this he always thereafter greeted me as “that allergist from Leeds”!! I also visited him some years later and again found him a very freindly, intersting and stimulating person.
1987 Hodson ME, Roberts CM, Butland RJA, Smith MJ, Batten JC. Oral ciprofloxacin compared with conventional intravenous treatment for Pseudomonas aeruginosa infection in cystic fibrosis. Lancet 1987; i: 235-7. [PubMed]
An early study of one of the first oral anti-Pseudomonal antibiotics, ciprofloxacin, showing benefit. Not surprisingly the oral treatment was preferred to IV treatment by 17 of the 20 ciproxin-treated patients! Aoral anti-Pseudomonal drug was a major advance in treatment – all previous systemic anti-Pseudomonal antibiotics had required either intravenous or intramuscular administration. Eventually, combined with inhaled colomycin, ciprofloxacin became the standard eradication treatment for early Pseudomonas infection (Valerius et al, 1991).
Undoubtedly an oral anti-Pseudomonas drug was a major advance in treatment. We also reported the successful use of another oral anti-pseudomonal antibiotic, Enoxacin, in a 12 year old boy with cystic fibrosis (Miller MG et al. Lancet 1985; i:646). There was some anxiety regarding the possibility of joint problems – “I don’t want bad joints like those puppies got!’ said one little boy.
1987 Beverley DW, Kelleher J, MacDonald A, Littlewood JM, Robinson T. Comparison of four pancreatic extracts in cystic fibrosis. Arch Dis Child 1987; 62:564-568. [PubMed]
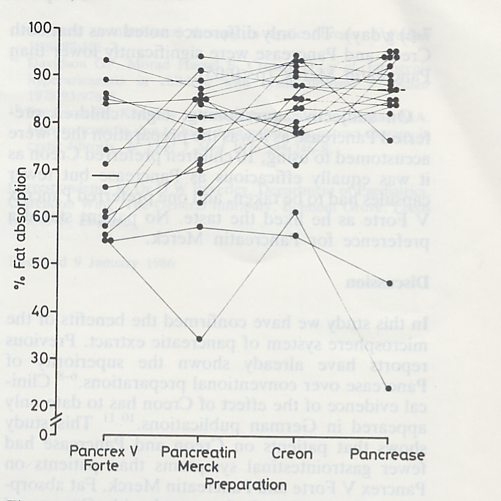
Fig 45. Percentage fat absorption vs. type/brand of enzyme. With permission.

Fig. 46 David Beverley
A controlled trial of ‘old’ (Pancrex V Forte) and new acid resistant enzymes (Pancrease, Creon, Pancreatin Merck – the last later marketed as Nutrizym GR). The study confirmed the marked superiority of the new preparations Pancrease and Creon that achieved lower abdominal symptom scores and significantly better median fat absorption (87% and 85%) and nitrogen absorption (faecal nitrogen of 1.6 and 2.1g/day) than the other two preparations Pancrex V Forte and Pancreatin Merck (Nutrizym) with fat absorptions of 74% and 81% and median faecal nitrogen of 2.9 and 2.7g/day (figure 45).
Dr David Beverley (figure 46) was working with us at St James when this study was done. He subsequently moved to York as a consultant paediatrician and developed the CF service there; I visited his excellent CF clinic every few months.
Undoubtedly the introduction of these new acid-resistant enzymes was one of the main advances in clinical care during the decade (also Weber et al, 1979 above; Khaw et al. 1977&1979 above; Holsclaw DS & Keith H, 1980 above; Gow et al, 1981 above; Mischler EH et al. 1982)
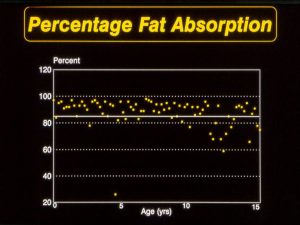
Fig 48 percentage fat absorption receiving full care at Leeds
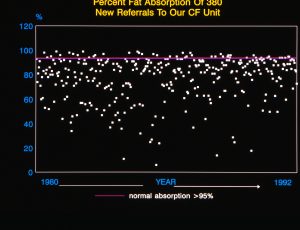
Fig 47 percentage fat absorption of 380 new referrals to St James’s 1980-1992
The percentage intestinal fat absorption of 380 new referrals to our CF Centre for assessment between 1980 and 1992 (Fig 47) shows that many patients had inadequate control of their intestinal malabsorption at the time of referral but with a tendency to improve (which when analysed was significant) in those referred in the later years when the new enzymes were increasingly already prescribed by referring consultants (figure 47). Fat absorption in Leeds patients where there was a particular interest in nutrition and monitoring and treatment of malabsorption (Fig.48).
1987 Pedersen SS, Jensen T, Hoiby N, Koch C, Flensborg EW. Management of Pseudomonas aeruginosa lung infection in Danish cystic fibrosis patients. Acta Paediatr Scand 1987; 76:955-961. [PubMed]
This is one of the main publications justifying the Danish policy of 3-monthly courses of intravenous antibiotics for patients chronically infected with Pseudomonas – a policy which has never been subjected to an acceptable clinical trial and as mentioned above was initiated before the advent of inhaled antibiotics. The annual mortality rate of cystic fibrosis patients with chronic P. aeruginosa lung infection at the Danish CF-centre ranged from 10 to 20% in the years 1970-1975 – in this period the patients received anti-Pseudomonal chemotherapy only during acute exacerbations of infection.
From 1976-1979 patients who acquired chronic P. aeruginosa infection were given regular and intensive anti-Pseudomonal treatment three to four times per year. The patients were followed for 6-12 patient-years; seven died and the 10-year survival rate after onset of P. aeruginosa infection was 90% (+/- 4%). The annual mortality rate is now only 1-2%. Although precipitating antibodies against P. aeruginosa increased significantly, pulmonary function did not deteriorate with duration of infection.
An unwelcome consequence of this policy was an increase in cross-infection between patients associated with more frequent hospitalisation and an increased incidence of new P. aeruginosa infection in 1976 which was steadily reduced, starting in the late Seventies, by improved hygienic measures and a new ward in the CF centre.
1987 Jensen T, Pedersen SS, Garne S, Heilmann C, Hoiby N, Koch C. Colistin inhalation therapy in cystic fibrosis patients with chronic Pseudomonas aeruginosa lung infection. J Antimicrob Chemother 1987; 19:831-838. [PubMed]

Fig 49. Tim Jensen & Tacjana Pressler. Author’s photo
One million units of inhaled colistin twice daily were compared with isotonic saline over three months in patients chronically infected with P. aeruginosa. More patients in the colistin group completed the study (18 vs.11). In terms of symptom scores, maintenance of pulmonary function (but only limited to the FVC) and inflammatory parameters, colistin was superior to placebo. The Tim Jensen and the Danish group (Jensen et al, 1987) (Fig 49) apparently started to use nebulised colistin on their chronically infected patients following our letter from Leeds to the Lancet reporting its use in eradicating early Pseudomonas infection (Littlewood et al, 1985 above).
Although the results in this present study were not impressive, inhaled colistin came into widespread use in the UK and Europe to stabilise patients with chronic Pseudomonas chest infection – as it turned out, on the relatively modest published evidence in this paper!
Eventually studies showed colomycin to be less effective for chronic infections than inhaled tobramycin (TOBI) but both treatments were definitely associated with a significant fall in the numbers of bacteria in the sputum (Hodson ME et al. Eur Respir J 2002; 20:658-664. [PubMed]. However, in this comparison the patients had received colistin previously but were TOBI (tobramycin) naive and hence were more likely to improve.
Dr Tacjana Pressler (Fig. 49) eventualy became Director of the Copenhagen CF Centre.
It is of interest that on relatively little published evidence that colomycin became the most widely used inhaled anti-Pseudomonal antibiotic in Europe and still remains widely used and it remains the mainstay of effective early P. aeruginosa eradication therapy. Treatments that work eventually come into use w
1987 Stead RJ, Davidson TI, Duncan FR, Hodson ME, Batten JC. Use of a totally implantable system for venous access in cystic fibrosis. Thorax 1987; 42:149-150. [PubMed]
One of the first reports of experience with this excellent device (Port-A–Cath) for adults with CF requiring frequent intravenous treatment but whose peripheral veins were becoming increasingly difficult to access (also McDowell et al, 1986 above; Essex-Cater et al, 1989 below).
1987 Penketh A, Higenbottam T, Hakim M, Wallwork J. Heart lung transplantation in patients with end stage lung disease. BMJ 1987; 295:311-314. [PubMed]

Fig 50. John Wallwork
The cardiothoracic surgical team at Papworth, Cambridge UK under the leadership of Mr John Wallwork (figure 42) report preliminary results of heart-lung transplantation in seven patients one of whom had CF. Six of the seven patients were well after four to 33 months. Papworth became one of the major UK transplant centres for people with cystic fibrosis.
Further experience was published in 1989 where five of 33 patients referred benefited over 18 months (Smyth RL et al. Arch Dis Child 1989; 64: 1225-1229. [PubMed] The first heart lung transplants for CF in the UK were performed by Mr. Magdi Yacoub in 1985 in London at Harefields Hospital (Yacoub et al, 1990 below).
1987 Penketh ARL, Wise A, Mearns MB, Hodson ME, Batten JC. Cystic Fibrosis in adolescents and adults. Thorax 1987; 42:526-532. [PubMed]

Fig 51 Andrea Penketh
This is a report of the extensive experience of adults with CF at the Brompton Hospital in London, one of the first and still the largest adult CF centre in the world.
Three hundred and sixteen patients with CF had attended the Brompton Hospital during 1965-83; 178 (56.3%) of them were male and 136 female, and their ages ranged from 12 to 51 years. Most patients presented in infancy with respiratory symptoms and malabsorption, but 19 (6%) were diagnosed in adult life, three in their thirties. Pulmonary disease was almost universal (99.7%), being responsible for 97% of all deaths and three quarters of hospital admissions. All patients had developed a productive cough by the age of 21 and over half before the age of 5 years. Many complained of wheezing and reversible airflow obstruction was present in 40% of those tested. Minor haemoptysis was very common (62%), but major episodes less so (10%). Pneumothorax was seen in 61 cases (19%), and was often recurrent. Some irreversible airflow obstruction was present in all patients with pulmonary disease. Two patients had been followed for over 20 years without showing appreciable decline in lung function. Thirty five patients (11%) had no symptoms of malabsorption. Acute meconium ileus equivalent was seen in 16% and a chronic partial obstruction with episodic symptoms in a further 19%. Diabetes mellitus developed in 36 patients, 13 of whom were insulin dependent. Hepatomegaly was common (29%), often occurring without abnormal results in biochemical tests of liver function; only 1% of patients developed portal hypertension with varices and ascites. Skin reactions to at least one common allergen, including Aspergillus fumigatus, were positive in 70%, but very few patients suffered from hay fever or eczema. One hundred and twenty one patients have died, 97% from infection or other pulmonary complications, and 195 were alive in December 1983 (mean age 23 years). Seventy eight per cent of patients were in full time education or full or part time employment, or were housewives, and only 41 were unemployed for reasons for health. Many patients were married and 10 women had borne children. Most patients were admitted to hospital only three or four times during the period of follow up and 50 individuals (16%) had never been in hospital at all.
The authors suggested that the improvement in prognosis and quality of life for adults with cystic fibrosis should encourage a positive attitude in those who care for them.
A detailed account of experience from 1965 at the Brompton Hospital, London which was and still is the largest centre in the world for adults with CF – hence the data is summarised here in some detail. The clinic was started by Sir John Batten in 1965 and attracted patients from near and far – many coming from Archie Norman’s clinic at Great Ormond Street, London or from Margaret Mearns and Winifred Young at the Queen Elizabeth Hospital Hackney, London – at both these paediatric clinics the treatment was of a higher than the average standard in the rest of the UK.
Dr Andrea Penketh (figure 51) is consultant in General and Respiratory Medicine at Cheltenham. Her special interests include Cystic Fibrosis following a number of research publications and an MD thesis undertaken at the Brompton Hospital in 1980s. Her MD thesis on the behaviour of Pseudomonas aeruginosa was in Cystic Fibrosis. She has over 30 publications on Cystic Fibrosis and respiratory medicine including the early description of the use of nebulised antibiotics in chronic P. aeruginosa infection in CF and the use of bronchial biopsy to diagnose rejection in lung transplantation.
1988 Reisman JJ, Rivington-Law B, Corey M, Marcotte J, Wannamaker E, Levison H J. Role of conventional physiotherapy in cystic fibrosis. Pediatr 1988; 113:632-636. [PubMed]

Fig. 52 Joe Reisman med.uottawa.ca
A frequently quoted paper from Toronto of a 3-year prospective study to compare the long-term effects of postural drainage accompanied by percussion and the forced expiratory technique with the effects of the forced expiratory technique alone. Patients who performed the forced expiratory technique alone had mean annual rates of decline that were significantly different from zero for forced expiratory volume in 1 second (p <0.001), forced expiratory flow between 25% and 75% of vital capacity (p <0.001), and Shwachman clinical score (p< 0.004). In the group performing conventional physiotherapy with percussion and postural drainage, only the mean annual rate of decline for forced expiratory flow between 25% and 75% of vital capacity was significantly different from zero (p<0.03), and it was significantly different from the mean rate of decline associated with the forced expiratory technique alone (p<0.04). The authors concluded that conventional chest physiotherapy should remain a standard component of therapy in cystic fibrosis.
Physiotherapy is a major and time-consuming component of therapy and the most irksome part of treatment for many patients and parents. This frequently quoted study was one of the few to demonstrate that the time was well spent and so the paper was useful and was quoted widely for this reason (also Pryor et al, 1979 above; also Desmond et al, 1983; Tyrell et al, 1986 below).
Dr Joe Reisman (Fig. 52) qualified in Toronto where, after extensive training appointments, he eventually worked in various senior clinical and academic roles. His involvement with training was recognised by two awards from the Lung Association. In 2000 he became Chief of Paediatrics at the Children’s Hospital of Eastern Ontario, Ottawa and Chair of Paediatrics at the University of Ontario.
1988 Buchdahl RM, Cox M, Fulleylove C, Marchant AL, Tomkins AM, Brueton MJ, Warner JO. Increased resting energy expenditure in cystic fibrosis. J Appl Physiol 1988; 64:1810-1816.[PubMed]

Fig 53. Roger Buchdahl
Roger Buchdahl (fig. 53) working at the Brompton Hospital measured resting energy expenditure (REE) by indirect calorimetry in 23 subjects with CF in a stable clinical state and in 42 normal control subjects. In subjects with CF the REE was found to be elevated by an average of 9.2% above expected values derived from the control subjects. There were significant correlations between increased values and poor pulmonary function. However; the correlations were low, suggesting that other factors may contribute to the increased resting energy expenditure, possibly including the putative metabolic defect in cystic fibrosis.
This study provided confirmation of previous original observations from Toronto (Pencharz et al, J Pediatr Gastroenterol Nutr 1984; Suppl 1:S147-53 above; Vaisman et al, J Pediatr 1987; 111:496-500. [PubMed]) that most children with CF have increased resting energy expenditure which appears to be related to and correlated with the severity of the chest infection. Also CF infants have some 25% greater energy expenditure than controls (Shepherd RW, et al. Lancet 1988; i: 1300-1303. [PubMed]; also Bowler et al, 1993 below).
Dr Roger Buchdahl (Fig. 53) was later consultant paediatrician at Hillingdon Hospital and the Brompton Hospital, London.
1988 Stringer DA, Sprigg A, Joudis E, Corey M, Daneman A, Levison HJ, Durie PR. The association of cystic fibrosis, gastroesophageal reflux and reduced pulmonary function. Can Ass Radiol J 1988; 39:100-1002.[PubMed]
Between 1971 and 1984 fifty seven patients with CF had a barium meal for suspected gastroesophageal reflux which was found to be present in 18 (32%) of those examined – in six the reflux was associated with a hiatus hernia, oesophagitis or stricture. The 18 patients with reflux had significantly worse respiratory function than the other 412 people with CF in the clinic without known reflux.
In a number of subsequent publications gastroesophageal reflux was shown to be a frequent and significant problem in both children and adults with CF and other chest problems. The complication was first described by Jean Feigelson (Feigelson J, Sauvegrain J. Reflux gastroesophagien dans la mucoviscidose. N Press Med 1975; 4:2727-2730 above). Other notable contributions included Malfroot A, Dab I. New insights of gastro-oesophageal reflux in cystic fibrosis by longitudinal follow-up. Arch Dis Child 1991; 66:1339-1345 below; Ledson MJ et al. Prevalence and mechanism of gastro-oesophageal reflux in adult cystic fibrosis patients. J R Soc Med 1998; 91:7-9 below).
More recently the association of reflux with head down postural drainage manoeuvres in infants with CF has altered the practice of many physiotherapists by avoiding the head down position (Button BM et al. Postural drainage and gastro-oesophageal reflux in infants with cystic fibrosis. Arch Dis Child 1997; 76:148-150 below); however, not all would accept these findings regarding the head down position.
1988 Gaskin KJ, Waters DL, Howman-Giles R, de Silva M, Earl JW, Martin HC, Kan AE, Brown JM, Dorney SF. Liver disease and common-bile-duct stenosis in cystic fibrosis. N Engl J Med 1988; 318:340-346. [PubMed]
This study from Sydney caused considerable interest. Hepatobiliary scans were performed on 50 of 61 patients with CF who had hepatomegaly, abnormal liver function, or both and also in 31 of 92 patients with CF who did not have hepatomegaly or abnormal liver function. Ninety-six percent of the patients with liver disease had evidence of biliary tract obstruction with a stricture of the distal common bile duct in the majority of cases. All the patients without liver disease had normal intra hepatic and common-duct excretion of tracer. The authors concluded that strictures of the distal common bile duct are common in patients with CF and liver disease. They said the association required further study, since surgical relief of common-duct obstruction may prevent or ameliorate the hepatic complications of cystic fibrosis.
If strictures of the common bile duct were an important cause of CF related liver disease this was a very important observation. However, the results were questioned and the findings were contested in later publications for example by Nagel RA, Westaby D, Javaid A, et al. Liver disease and bile duct abnormalities in adults with cystic fibrosis. Lancet 1989; ii: 1422-1425. [PubMed]. Brien S, Keogan M, Caseu M, et al. Biliary complications of cystic fibrosis. Gut 1992; 33:387-391). [PubMed] concluded “these results indicate that abnormalities of the bile ducts in patients with cystic fibrosis related liver disease are confined to the intrahepatic biliary tree and that common bile duct strictures do not contribute to either the progression or development of liver disease in these patients”.
Subsequently bile duct obstruction was not established or considered as an important cause of CF related liver disease.
1988 Gibbens DT, Gilsanz V, Boechat MI, Dufer D, Carlson ME, Wang CI. Osteoporosis in cystic fibrosis. J Pediatr 1988; 113:295-300. [PubMed]
Although osteoporosis had been noted in a number of previous publications, this was one of the early papers mainly dealing with osteoporosis, a complication which would become an increasingly common problem as the average age of people with CF increased.
Vertebral bone density of 57 CF patients (aged 3-21 yrs) was compared with 57 matched controls. The bone density in people with CF was 10% less than in controls and worse in those with in a poor nutritional state. (For earlier mention of osteoporosis Mischler et al, 1979 above).
1988 Wood RE, Piazza F. Survival in cystic fibrosis: correlation with treatment in three cystic fibrosis centres. 10th International Cystic Fibrosis Congress, Sydney 1988 Excerpta Medica Asia Pacific Services 74:79.
Fig 54 Robert Wood
An impressive and important, if disturbing, study presented as a poster at the 1988 Sydney CF meeting. The medical records of three US CF Centres for the years 1971-1980 were reviewed including 24,000 outpatient visits and 2000 hospitalisations. Median survival ages at the three centres were 9.5, 18.1 and 22.8 years for centres A, B and C respectively. No differences in the content of treatment between centres correlated with group survival. But for each patient year in A, B and C the mean number of outpatient visits was 4.4, 8.0 and 12.1 and the number of hospital days was 4.7, 5.0 and 9.5. – usually hospitalisations would be for IV antibiotics. Hospitalisation was considered necessary in 19.3, 20.9 and 31.2% of patients at least once a year. There was strong correlation between group survival and the use of oral and aerosolized antibiotics, vitamins E and K, and aerosols of epinephrine. When the data was separated into 1971-1975 and 1976-1980, changes in antibiotic use correlated well with improved survival.
The authors concluded “more frequent outpatient visits, more aggressive use of antibiotics and more frequent hospitalisation correlate well with increasing survival”.
It is disappointing that this excellent study was never published. I suspect the marked differences between the CF centres were unacceptable and the data too sensitive for publication at that time.
Interestingly similar studies, some very recent, show patients are better when seen more frequently and receive more antibiotics – which is exactly what one would expect!
1988 Gilbert J, Robinson T, Littlewood JM. Home intravenous antibiotic treatment in cystic fibrosis. Arch Dis Child 1988; 63:512-517. [PubMed]

Fig 55. John Gilbert

Fig 56. Teresa Robinson
An early study of home intravenous antibiotic therapy for CF children showing that, with adequate support, home IV was as effective as hospital treatment, and preferred by families and considerably less expensive.
A subsequent unpublished study showed that 2 weeks of home IV antibiotics cost approximately £1.5K and the same treatment in hospital £2.5K.
In this present paper our CF Research Fellow John Gilbert (Fig. 55) describes our experience during the first 20 months of using a system of home intravenous antibiotic treatment in which our cystic fibrosis liaison nurse (Teresa Robinson. fig. 56) had a central role. Thirteen patients received 40 courses of treatment. There were highly significant improvements in weight, respiratory function, and white cell count during home treatment. There was no significant difference in weight and forced expiratory volume in one second between the end of home treatment and the end of hospital treatment while forced vital capacity was better after home treatment. All patients preferred home treatment. The advantages of home visits by the CF liaison nurse during treatment were emphasised.
Subsequently numerous studies were published attesting to the feasibility, effectiveness and patient acceptability of home intravenous antibiotic therapy – an obvious major advance in treatment. However, surprisingly, a Cochrane Review in 2000, updated in 2006, considered only one small study as suitable for inclusion which showed home therapy did not harm the patients! Despite this lack of acceptable evidence for the Cochrane disciples, home IV therapy was undoubtedly one of the major advances of the decade and became a central feature of CF treatment. Failure of a CF Centre to provide this option for patients would be regarded as a major deficiency.
1988 Corey M, McLaughlin FJ, Williams M, Levison H. A comparison of survival growth and pulmonary function in patients with cystic fibrosis in Boston and Toronto. J Clin Epidemiol 1988; 41:588-591. [PubMed]

Fig 58. Mary Corey
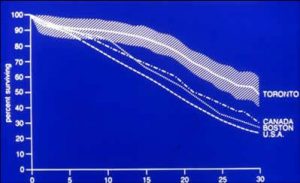
Fig 57. Survival graph from Corey et al. 1988
An oft-quoted study comparing survival curves between 1972-1981 of 499 Boston patients and 534 Toronto patients (fig. 57). The Toronto patients were taller and heavier than the Boston patients but had similar respiratory function. Median age of survival in Boston was 21 years and in Toronto 30 years with divergence occurring from the age of 10 years.
The authors concluded “The differences in growth and survival in these two patient groups, with very similar age specific pulmonary function, suggest further examination of nutritional guidance and intervention in CF especially regarding the traditional restriction of dietary fat”.
Douglas Crozier in Toronto had recommended a normal or high fat intake with very large doses of Cotazym since the early Seventies, at a time when many clinicians advised a fat-restricted diet (Crozier, 1974 above).
Mary Corey (fig. 58) has been CF Research Scientist at the Toronto Hospital for Sick Children since the Seventies and is co-author in over 150 publications from Toronto mostly on various aspects of cystic fibrosis. In 2007 she was co-Chairman of the North American Cystic Fibrosis Conference programme committee.
1988 MacDonald A, Kelleher J, Littlewood JM. A normal fat diet for cystic fibrosis: is a dietitian still needed? Scand J Gastroenterol 1988; 23 (Suppl 143):157-159. [PubMed]

Professor Anita MacDonald OBE
Facebook

Fig 59. Anita MacDonald
In this paper the dietary intakes of 90 people with CF who came to Leeds for Comprehensive Assessment; they had received various degrees of dietetic support at their local hospitals. They were assessed by a 7-day dietary history.
Patients who had contact with a dietitian at least twice a year achieved higher energy and protein intakes than patients with little or no dietetic help. Patients who had one intensive interview with a specialist CF dietitian but had little local dietetic support attained better nutrient intakes than patients with no dietetic support.
At that time dietary misconceptions, which still led to restriction of fat and even sugar, were still relatively common in the UK particularly when patients or parents of children with CF received little dietary advice. Only 8% of patients receiving no dietary advice achieved energy intakes of more than 120% of the recommended daily allowance compared with 50% of those seeing a dietitian – the difference being largely related to the fat intake. So it seemed a dietitian was still needed!
Anita MacDonald (fig. 59), at the time of this study was the senior dietitian at the Leeds Paediatric CF Centre at St James’s. She reported experience with patients referred to Leeds for our so-called Comprehensive CF Assessment. Anita made a major and quite exceptional contribution to the nutritional management of children with CF attending the Leeds centre and to the development of the regional CF service. Eventually, Anita wanted more involvement with metabolic conditions and moved as Chief Dietitian at the Birmingham Children’s Hospital but left a legacy of expertise in Leeds which was continued and further built on by Sue Wolfe and Alison Morton our two chief dietitians. Eventually Anita became a Professor at Birmingham was eventually awarded an OBE for her services to medicine.
1988 Littlewood JM, Kelleher J, Rawson I, Gilbert J, Firth J, Morton S, Wall C. Comprehensive assessment of patients at a CF centre identifies suboptimal treatment and improves management, symptoms and conditions. 10th International Cystic Fibrosis Congress, Sydney 1988 Excerpta Medica Asia Pacific Services. 89-90.
This poster from Leeds reported the findings of the first 250 new patients with CF referred to our Leeds unit between May 1980 and September 1987 for what we termed “Comprehensive CF Assessments” and advice. It was because we offered to carry out Comprehensive Assessments on other paediatricians’ patients, without taking over the care of the majority of the patients, that the service we offered developed so rapidly during the Eighties.
During two day-long visits to the Leeds Regional CF Unit the patients underwent a detailed review of their past history, symptoms, treatment, physical state and diagnosis. There was a confirmatory sweat test, assessment by the dietitian and physiotherapist , respiratory function tests chest and abdominal X-rays and abdominal ultrasound. Sputum culture, detailed haematology, biochemical and immunological blood tests including fat soluble vitamin levels and a faecal fat and chymotrypsin estimation were performed.
The day finished with an hour with the CF consultant (Jim Littlewood) who by then had considerable detail about the patient available including the referral letter from their paediatrician, the extensive data obtained by our permanent CF clinic doctor, nurses, dietitian, physiotherapist, respiratory function tests and chest and abdominal x-rays (also Littlewood et al, 1984 above; Littlewood et al, 1993 below).
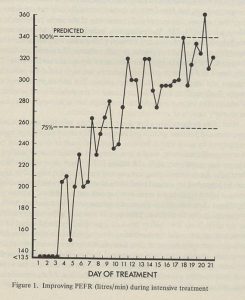
Fig 61. Respiratory function (peak expiratory flow rates) same child a Fig. 59during course of IV antibiotic treatment
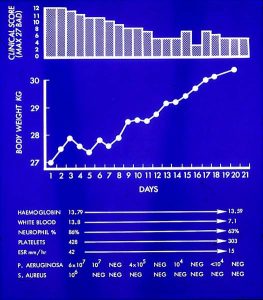
Fig 60. Clinical progress of a typical child referred at that time
Both the treatment and the physical state were frequently found to be suboptimal and, it must be said, reflected treatment in many general hospitals in the UK during the Eighties.
For example, of our first 250 referred patients, the diagnosis was incorrect in 7 (3%), the chest was obviously under treated in 30%, physiotherapy was suboptimal or not preformed at all in 60%, the energy intake was inadequate and less than 120% of the recommended daily allowance in 75%, pancreatic supplements inadequate in 40% and vitamin supplements insufficient resulting in suboptimal plasma levels in 60%.
An typical example of some of the patients referred at this time (figs. 60 and 61). A girl of 12 years was admitted on the day she came for a Comprehensive Assessment as the state of her chest was so bad. She came with the firm encouragement of the Director of the CF Trust who knew the family. Before referral from her local hospital she had never had IV or nebulised antibiotics although she was chronically infected with Pseudomonas aeruginosa.
She was given 3 weeks intravenous anti-Pseudomonal antibiotics and physiotherapy with a dramatic response in clinical score, weight and laboratory findings (fig. 60) and respiratory function (fig. 61). This was the type of patient referred to as “under-treated.” Unfortunately, although such patients improve dramatically for a few years when treated, some then deteriorated despite treatment, as did this girl, who died aged 15 years.
1988 Jones K, Higenbottam T, Wallwork J. Successful heart-lung transplantation for cystic fibrosis. Chest 1988; 93:644-5. [PubMed]
Report of one of the first successful heart lung transplants in CF in October 1985 by Mr Wallwork’s team at Papworth Hospital, Cambridge. Sixteen months after heart-lung transplantation, the FEV1 of a young woman, who had been in the terminal stages of CF, has risen from 16 percent (0.6 L) to 77 percent of her predicted value. She had returned to work.
These first results of heart lung transplantation for a condition which was, up to that time, uniformly fatal were quite dramatic and excited a great deal of hope and interest in the CF community. John Wallwork and Magdi Yacoub were leaders in this field in the UK for the next few years. (also Penketh et al above; Scott J et al. Heart lung transplantation for cystic fibrosis. Lancet 1988; ii: 192-194 reporting five of six patients with CF survived heart lung transplantation at Papworth Hospital).
Later Prof John Dark and Prof Paul Corris in Newcastle developed a major transplant service in the UK.

Fig. 63 Norman Shumway Westernthoracic.org

Fig. 62 Bruce Reitz Doximity
The world’s first successful heart-lung transplant was carried out at Stanford by cardiothoracic surgeon Bruce Reitz (Fig.62) in March 1981. The surgical team included Norman Shumway (Fig. 63) (1923-2006), Philip Oyer and many others. Reitz had been working with Norman Shumway, who had performed the first heart transplant on an adult in 1968, for some years. Forty five year old Mary Gohike, who received the donor organs of a teenage killed in a traffic accident, recovered well and lived for a further 5 years.
1988 Simmonds EJ, Mahony MJ, Littlewood JM. Convulsion and coma after intranasal desmopressin in cystic fibrosis. BMJ 1988; 297:1614. [PubMed]

Fig. 62 Eddie Simmonds
A 13 year old girl with CF admitted for treatment of an exacerbation of her chest infection also had nocturnal enuresis for which she was treated with desmopressin spray. This was given for 4 nights -10ug, 20ug, 20ug and 10ug. After the fourth dose she was noted to have gained weight, developed severe hyponatraemia and had a major convulsion lasting 4 minutes after which she remained comatose and passed no urine for 28 hours. She gradually regained conciousness over 2 weeks and returned to normal with no neurological sequelae. We were not clear as to why this severe reaction occured although suggested that it was in some way linked to the chloride channels abnormalities.
Dr Eddie Simmonds (fig. 62) was our CF Research Fellow at St. James’s at the time of this report. Eddie subsequently was appointed consultant paediatrician in Coventry where he also built up a successful CF clinic.
1988 Brown GA, Sule D, Williams J, Puntis JWIL, Booth IW, McNeish AS. Faecal chymotrypsin: a reliable index of exocrine pancreatic function. Arch Dis Child 1988; 63:785-789. [PubMed]
Specimens of meconium and random stools were collected sequentially from 25 healthy newborn babies over the first eight to 14 days of life. The stool chymotrypsin concentrations increased from birth to a maximum at four days of age and then fell again over the next four days. In 22 newborn babies suspected of meconium ileus and later confirmed to have CF, faecal chymotrypsin concentrations were all appreciably reduced. Measuring faecal chymotrypsin concentrations was a reliable procedure for identifying pancreatic exocrine insufficiency in the newborn. (also from Birmingham, Brown GA et al. Arch Dis Child 1988; 63:1229-1233). [PubMed]
This test is very useful in management of CF patients and I have described the use of the test recently as follows – “occasional measurements of faecal chymotrypsin are useful for monitoring pancreatic enzyme replacement therapy. Values that remain low on treatment indicate either inadequate prescription or non adherence with treatment although a normal value does not exclude persisting significant steatorrhoea. Very high values can indicate that the dose is excessive. High values also occur if the patient has rapid intestinal transit. Conversely constipation may be associated with low values. If low values persist alongside symptoms of malabsorption treatments to reduce gastric acid secretion may help improve enzyme efficacy and enable reduction in enzyme intake (Littlewood et al. Pediatr Pulmonol 2006; 41:35-39. [PubMed]).
Unfortunately, the test was subsequently withdrawn by some UK laboratories when faecal elastase 1 became available as a marker of pancreatic function.
1988 Dodge JA, Goodall J, Geddes D, Littlewood JM, Mearns MB, Owen JR, Russell G. Cystic fibrosis in the United Kingdom 1977-85: an improving picture. British Paediatric Association Working Party on Cystic Fibrosis. BMJ 1988; 297:1599-1602. [PubMed]
A further publication from the Working Party on Cystic Fibrosis chaired by Prof. John Dodge. The national survey included all patients with CF known to paediatricians, chest physicians and others between 1977 and 1985 or whose deaths were reported through death certification authorities in the United Kingdom. The number indicated an incidence of 1 in 2500 live births. Mortality was 7.6% in the first year and greater for females than males under 20 years of age. Survival improved through the study particularly in the first five years with 100 more births than deaths each year. In particular, death from meconium ileus was increasingly less frequent. It was estimated that there were some 5000 people with CF in 1985. (1982 Formation of UK CF Working Party, above; Jackson 1986 for summary of the initial Working Party report, above; Dodge et al, 2007 final data, below).
1988 Littlewood JM, Johnson AW, Edwards PA, Littlewood AE. Growth retardation in asthmatic children treated with inhaled beclomethasone diproprionate. Lancet 1988;865.[PubMed]
This was the first report of an adverse effect on growth of some children with asthma receiving inhaled corticosteroids. It is mentioned here as many children with CF receive inhaled steroids often in very large doses in an effort to control their symptoms. Graff-Lonnevig V et al 1979. [PubMed]) had reported inhaled steroids had no effect on growth – surprisingly even though the height SDs fell slightly (but “insignificantly”) in the children receiving inhaled steroid treatment. Also Simon Godfrey also had found no adverse effect on growth (Godfrey et al. 1978. [PubMed]).
However, during the Eighties there were 346 asthmatic children who attended my asthma clinic at St James’s University Hospital in Leeds at least once between April 1984 and March 1985. They were accurately weighed and measured (using a Harpenden Stadiometer) by the experienced clinic nurses at every attendance. I noticed that after inhaled steroids were started some children had an increase in weight and an impressive reduction in asthma symptoms but, unexpectedly, had no improvement or even a fall off of height growth; one would have expected their height to be accelerating as their asthma improved with the treatment.
Fig. 63 Typic fall off in height gain after starting inhaled steroids
So we analysed the growth of all the children with asthma who were receiving inhaled steroids in the clinic (fig. 63) When the beclomethasone (BDP) group were compared with controls their SDS for height and weight were lower than controls. Of 16 patients studied before and after starting BDP treatment (figure 61) there was a significant negative inflection of growth pattern closely related to the start of BDP treatment – this demonstrated a definite adverse effect on height.
We concluded that “Although it has been suggested that relevant clinical side effects from inhaled steroids are virtually non-existent, our data in children treated with inhaled steroids does not support this view”.
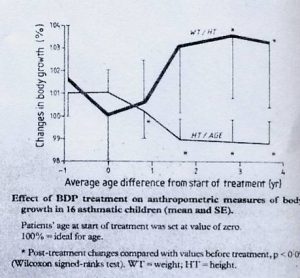
Fig 63. height before and after starting inhaled steroids. With permission/
This observational study came in for heavy criticism from some respiratory paediatricians!! Over some 15 years they had failed to identify the adverse effect on growth (Godfrey S, et al. J Allerg Clin Immunol 1978; 62:335-339.)[PubMed] or had attributed the fall off in growth rate entirely to the delay in puberty (e.g. Balfour-Lynn L. Effect of asthma on growth and puberty. Pediatrician 1987; 14:237-41. [PubMed]
For example Balfour-Lynn states – “The main cause of growth retardation in the past was the long-term prophylactic use of oral corticosteroids. Since the advent of inhalation steroids, this has no longer been a problem”.
However, some of our Leeds children were only 7 and 8 years old when they experienced an obvious slowing of height gain at a time when their asthma was under better control.
Subsequently our observations on the adverse effect inhaled steroids on growth were confirmed in other studies although the controversy continued as, for many years, all were not convinced.
On reflection it seemed that many of the previous clinical trials were of too short duration to reveal side effects such as adverse effects on growth. It was apparent that even observational studies from large clinics by experienced clinicians, who were personally involved in long term patient care, were still important to reveal long term side effects. The basic requirement to reveal these problems appeared to be many patients accurately followed up by fewer experienced doctors, over longer periods of time and accurately measured by careful dedicated outpatient nurses. This paper and, as it turned out, the quite inappropriate, even scathing criticism it received, is reviewed in more detail on my personal website (jimlittlewood.com/the second of three original observations).
1989 Handyside AH, Pattinson JK, Penketh RJ, Delhanty JD, Winston RM, Tuddenham EG. Biopsy of human preimplantation embryos and sexing by DNA amplification. Lancet 1989; 1 (8634):347-349. [PubMed]

Fig 64 Alan Handyside
The first biopsy of a rabbit embryo had been performed in 1968 (Edwards RG & Gardner RL New Scientist 1968; 38:218-2). In this present paper Handyside and colleagues from the Hammersmith Hospital, London, reported the first unaffected child born following preimplantation genetic diagnosis for an X-linked disorder. A single cell was removed through a hole in the zona pellucida from each of 30 human embryos at the 6-10 cell stage three days after in vitro fertilisation. A normal proportion developed (37%) to the blastocyst stage and six hatched from the zona. Each male embryo was sexed from DNA amplification of a repeated sequence specific for the Y chromosomes. In 15 embryos with the normal two pronuclei the sex was also determined by in situ hybridisation.
Although at this stage not performed for CF, this technique of pre-implantation genetic diagnosis, was to prove a major advance for some CF carrier couples at high risk of having a CF infant, as it provided an alternative to prenatal diagnosis and termination if the fetus was affected – a course of action which was understandably unacceptable to many couples. The technique was first used for CF in 1992 (Handyside et al, 1992 below).
1989 Rayner RJ, Tyrell JC, Hiller EJ, Marenah C, Neugebauer MA, Vernon SA, Brimlow G. Night blindness and conjunctival xerosis due to vitamin A deficiency in cystic fibrosis. Arch Dis Child 1989; 64:1151-1156. [PubMe
Fig 65. Rosie Rayner
Rosie Rayner (fig. 65) then the CF Research Fellow at Nottingham and subsequently paediatrician in Wolverhampton, studied 43 patients with CF aged 8 to 44 years (median 16 years), for evidence of vitamin A deficiency. Eight had abnormal dark adaptation tests and three had conjunctival xerosis. Serum vitamin A and retinol binding protein concentrations were significantly lower in the affected patients who were also more likely to have abnormal liver function tests. Five patients were treated with 100,000-200,000 IU water miscible vitamin A orally and their daily vitamin supplements were increased to maintain normal concentrations. In four patients dark adaptation tests were repeated. Three were normal, but one patient required three further doses of water miscible vitamin A and a daily supplement of 12,000 IU vitamin A before her dark adaptation threshold returned to normal. Adolescents with cystic fibrosis are liable to develop night blindness and conjunctival xerosis, particularly if they have liver disease or fail to take daily vitamin supplements.
One of the papers reporting objective evidence of the clinical affects vitamin deficiency rather than merely reduced plasma levels. Obviously, very important if adults with CF, of which there were increasing numbers, were driving at night. Histological evidence of vitamin A deficiency was evident in many of the early post-mortem studies – in fact, for some time vitamin A deficiency secondary to intestinal malabsorption was regarded by Dorothy Andersen as a major factor in the pathogenesis of the condition.
1989 Devlin J, Beckett NS, Davd TJ. Elevated sweat potassium, hyperaldosteronism and pseudo-Bartter’s syndrome: a spectrum of disorders associated with cystic fibrosis. J R Soc Med 1989; 82 (Suppl 16):38-43. [PubMed]
Fig 66. Pseudo-Bartter’s syndrome. From Devlin et al. 1989
Three infants with “pseudo-Bartter’s syndrome” (the term first used by these authors) and two further with related electrolyte disturbances with low potassium and sodium and alkalosis (fig.66). They emphasise the importance of measuring the serum electrolytes at diagnosis and when unwell. They mention the first report in CF was by Rendle Short J (Arch Dis Child 1956; 31:28-30.above). [PubMed]
Bartter’s syndrome was described by Fred Bartter in 1962 (Bartter FC et al. Hyperplasia of the juxtamedullary complex with hyperaldosteronism and hypokalemia alkalosis. Am J Med 1962; 33:811-128 [PubMed]).
In 1978 we described successful treatment of a non-CF infant with Bartter’s syndrome using indomethacin (Littlewood JM, et al. Treatment of Bartter’s syndrome in early childhood with prostaglandin synthetase inhibitors. Arch Dis Child 1978; 53:43-48 [PubMed]]. Of course, children with CF who develop this syndrome require only salt replacement and an adequate sodium intake to remain well.
1989 Essex-Cater A, Gilbert J, Robinson T, Littlewood JM. Totally implantable venous access systems in paediatric practice. Arch Dis Child 1989; 64:119-123. [PubMed]
One of the first reports of successful use of totally implantable venous access devices (TIVAD) in UK children with CF; this followed a report of the successful use of these devices in children from the Liverpool paediatric oncology unit (McDowell et al, 1986 above).
Fig 68. devices with needles -Port-A-Cath on left, PAS Port on right
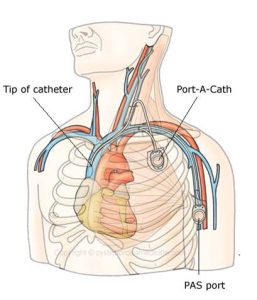
FIg 67. Port=A-Cath and PAS Port
This present study was a combined effort of the paediatric oncologists at St James University Hospital in Leeds (Dr Alison Essex-Cater, (fig. 69), who were already using the devices but had been somewhat discouraged by various serious complications including massive bleeding around the site. Our study was coordinated by our CF Research Fellow (John Gilbert.
The paper records our early experience with TIVADs and discusses the problems we encountered over the first three years. Forty seven TIVADs were inserted in 45 patients for the management of malignant disease (n = 29), haematological disorders (n = 5), and cystic fibrosis (n = 11). Subsequently TIVADs (fig, 68) became widely used in CF centres as intravenous antibiotic treatment increasingly became a major component of treatment.

Fig 69 Alison Essex-Cater
It became apparent that a surgeon with experience in their insertion was essential to minimise complications. Meticulous management to avoid infection of the device was important – often better given by conscientious well-trained parents than by inexperienced overworked medical and nursing staff! The teaching about TIVADs and their management became the responsibility of the CF Nurse Specialist who was also closely involved in organising home intravenous antibiotic therapy.
1989 Hill S, Phillips A, Mearns M, Walker-Smith JA. Cows’ milk sensitive enteropathy in cystic fibrosis. Arch Dis Child 1989; 64:1251-1255. [PubMed]
Over 12 years, proximal small intestinal mucosal biopsies were carried out in children with CF who had diarrhoea and failed to thrive in spite of adequate treatment, including pancreatic supplements. Histological examination of eight of the 17 biopsies taken over a period of 12 years from children with CF showed evidence of enteropathy, and accounted for one in 13 (8%) children with cystic fibrosis under 3 years of age attending Margaret Mearns’s CF clinic. Seven children clearly responded to a cows’ milk free diet; the diarrhoea stopped and weight gain increased. One of these responded only when gluten was also excluded from his diet. The eighth child remained on a normal diet and his symptoms did not improve. The enteropathy had resolved in all five patients who had further biopsies taken while receiving treatment, and from 15 months to 3 years of age all the children tolerated a normal diet and continued to thrive.
Cows’ milk sensitive enteropathy is an important cause of failure to thrive in a minority of children with cystic fibrosis – usually not revealed by allergy tests such as IgE or RAST. Small intestinal biopsy is an important investigation in younger children with CF who fail to thrive and have diarrhoea despite adequate treatment for they may have cow’s milk intolerance or even coeliac disease.
Prof. John Walker–Smith, originally from Australia, was Professor of Paediatric Gastroenterology at the Royal Free Hospital, London and a pioneer in the development of paediatric intestinal biopsy.
1989 Mieles LA, Orenstein D, Teperman L, Podesta L, Koneru B, Starzl TE. Liver transplant in cystic fibrosis. Lancet 1989; i: 1073. [PubMed]
This is the first report of liver transplantation in CF and the results are surprisingly good. Lung function, far from deteriorating as a result of the long operation has improved in some patients (Noble-Jamieson G et al, 1994. [PubMed] Successful heart-lung-liver transplantations (Noble-Jamieson et al, J R Soc Med 1996;89 (Suppl 27):31-37.) and lung-liver transplantations have also been performed (Couetil et al, 1995. [PubMed]; Couetil et al, 1997). [PubMed]
Fig 70. Liver with severe nodular cirrhosis removed at time of transplant
Figure 70 shows the liver removed from a 12 year old boy in liver failure. This was the first CF patient who had a liver transplant in Leeds, by the late Professor Geoff Giles, soon after this first paper appeared from Starzl’s team in the United States. In fact, I rang the Starzl team and received helpful advice and encouragement before we decided on this transplant. Our patient did very well after the operation.
Fig 71. Thomas Starzel
After 18 years in Denver, Thomas Starzl (1926-2017) (figure 71) accepted an appointment as Professor of Surgery at University of Pittsburgh, where he soon built the world’s largest liver transplant program. He also served as chief of transplantation services at Presbyterian University Hospital — now known as University of Pittsburgh Medical Center – and at the city’s Children’s Hospital and Veterans Administration Hospital. In 1985, he founded the Pittsburgh Transplantation Institute, the largest transplant program in the world. Over the years, Dr. Starzl has received most of the major awards of the medical profession and been decorated by the governments of many countries. He is among the few Americans ever inducted into the National French Academy of Medicine. In 2004, President George W. Bush awarded him the Presidential National Medal of Science.
1989 Choonara IA, Winn MJ, Park BK, Littlewood JM. Plasma vitamin K1 concentrations in cystic fibrosis. Arch Dis Child 1989; 64:732-734. [PubMed]

Fig. 72 Imti Choonara RCPCH
One of the first studies on vitamin K from the Leeds CF centre when Prof.(then Dr) Choonara (Fig. 72) worked there. Plasma concentrations in 37 patients mean age 10.6 years (2-23yrs) median 46ng/l and 16 controls 49ng/l. No relation found between an increase in prothrombin time and vitamin K plasma concentration.
However, subsequent studies using more efficient tests showed vitamin K was indeed deficient in many patients and even possibly related to later osteoporosis (Conway et al, 2005 below); vitamin K supplements are now recommended.
Prof. Imti Choonara (fig. 72) received the James Spence medal of the Royal College of Paediatrics and Child Health in a 2022. Described as “the founding editor of BMJ Paediatrics Open, a nationally and internationally renowned paediatrician and pharmacologist and a friend and supporter of child health and paediatricians around the world, particularly in Cuba where he has volunteered for many years”.
1989 Sokol RJ, Reardon MC, Accurso FJ, Stall C, Narkewicz M, Abman SH, Hammond KB. Fat-soluble-vitamin status during the first year of life in infants with cystic fibrosis identified by screening of newborns. Am J Clin Nutr 1989; 50:1064-1071. [PubMed]

Fig 72. Ronald Sokol
Another report on infants diagnosed in the Colorado neonatal screening programme. Fat-soluble vitamin status during the first year of life in 36 infants with CF was examined; biochemical evidence of fat-soluble-vitamin deficiency is common before the age 3 months in CF infants.
A further report on these 36 infants (Sokol RJ, et al. Pediatr Pulmonol 1991; Suppl 7:52-55.) after treatment with pancreatic enzymes, a multiple vitamin preparation, and additional vitamin E was associated with normalization of serum albumin, retinol, and 25-hydroxyvitamin D and negative PIVKA testing (for vitamin K) at six and 12 months of age. Several patients remained vitamin E deficient, but this was considered to be due to poor adherence to the treatment.
These studies from Denver of some of the earlier screened CF infants were important as they drew attention to the very early onset of nutritional deficiencies in the infants. Biochemical evidence of fat-soluble vitamin deficiency is common before three months of age and responds to adequate supplementation in the first year of life. Also the initial fall off in weight gain may take many months to recover.
Dr Ronald Sokol (fig. 72) is a paediatrician specialising in gastroenterology and hepatology at the Children’s Hospital Colorado and is a Professor of Pediatrics at the University of Colorado. He has published extensively on paedaitric gastroenterology, hepatology and drug abuse and in the present context has been involved with CF related liver disease and nutritional problems.
1989 Valletta EA, Mastella G. Incidence of celiac disease in a cystic fibrosis population. Acta Paediatr Scand 1989; 78:784-785. [PubMed]
Since the first report of coexistence of coeliac disease (CD) and CF in the same patient (Hide & Burman, 1969 above), isolated reports appeared of children with the two conditions. These authors report fivepatients with CD in a CF population of 1100 subjects – an incidence of 1 in 220. The most recent report is of the two conditions in a 56 year old man (Lampert S et al, Zeitschrift fur Gastroenterolgie 2007; 45:612-614). Cystic fibrosis has been shown to be risk factor for the development of coeliac disease which was present in 2.13% of patients (Walkowiak J et al. Acta Biochem Pol 2010; 57:115-118). [PubMed]
1989 Smith AL, Ramsey BW, Hedges DL, Hack B, Williams-Warren J, Weber A, Gore EJ, Redding GJ. Safety of aerosol tobramycin administration for 3 months to patients with cystic fibrosis. Pediatr Pulmonol 1989; 7:265-271. [PubMed]

Fig 73. Arnold Smith
Arnold Smith (fig.73) had noted tobramycin in urine samples after nebulised administration hence the present study to assess safety of 12 weeks of thrice daily inhalations of 0.6gm of preservative free tobramycin. There was no detectable laboratory evidence of nephrotoxicity, neither a decrease in auditory acuity (range 250-20,000 Hz) nor vestibular dysfunction. Pulmonary function tests significantly improved during the first month in all subjects but returned to enrolment values by the end of the 12th week of administration of the tobramycin aerosol. Sputum P. aeruginosa density initially decreased and remained significantly below the enrolment value throughout. Coincident with the reduced bacterial density, a reduction in cough frequency and sputum production, as well as a weight gain was observed. However, seventy-three percent of the patients with sputum P. aeruginosa isolates susceptible to tobramycin on enrolment yielded resistant organisms during aerosol administration although 1 year later all sputum P. aeruginosa isolates were susceptible to tobramycin. The authors concluded that thrice daily aerosol tobramycin administration for three months was safe although transient emergence of tobramycin resistant P. aeruginosa may occur.

Fig 74. Bonnie Ramsay
The favourable results of this study eventually led to the development, trial and introduction of TOBI – the tobramycin preparation specifically for inhalation – one of the major treatment advances of the Nineties (Ramsey BW et al. N Eng J Med 1999; 340:23-30 below).
Dr Arnold Smith. (figure 73) Professor, Department of Pathobiology, School of Public Health and Community Medicine, University of Washington primarily interested in infectious disease but has made major contributions to the antibiotic treatment of cystic fibrosis.
Dr Bonnie Ramsey (fig. 74) was honoured by the creation of the Bonnie W. Ramsey, M.D, Endowed Chair in Cystic Fibrosis, University of Washington in 2005. She is one of the leading figures in CF research and care in North America. Veteran CF clinician Dr Jack Docter of Seattle observed in 2002 that “by far my greatest contribution to the NCFRF and the CF cause was the recruitment of Dr Bonnie Watt Ramsey. Watching her career develop and observing her contribution to the field are two of the great satisfactions of my retirement” (In “Cystic Fibrosis in the 20th Century”. Carl Doershuk. 2002).
1989 Götz MH, Burghuber OC, Salzer-Muhar U, Wolosczuk W, Weissel M, Hartter E. Cor pulmonale in cystic fibrosis. J R Soc Med 1989; 82 Suppl 16:26-31. [PubMed]

Fig 75. Manfred Gotz
A detailed review of cor pumonale in cystic fibrosis by Manfred Götz of Vienna. They speculate that the problem will increase as the age of patients increases. Advances in the management of heart disease associated with chronic obstructive pulmonary disease may have relevance in relation to the complication in people with CF.
Professor Manfred Götz (fig.75) has been at the centre of CF research and clinical care in Europe since the Seventies and has published widely on many aspects of the condition, more recently collaborating with his wife, Ilse Götz, on various psychological issues.
1989 Fallon JS, Brocklebank JT, Simmonds EJ, Littlewood JM. Cystic fibrosis and renal tubular acidosis. Arch Dis Child 1989; 64::10054-5.[PubMed]
A case report of a child of consanguineous parents who had clinical features of two autosomal recessive conditions – cystic fibrosis and renal tubular acidosis.