
Nineties clinical


Steady improvement in clinical care and important new treatments
The improvement in outlook which characterised the Eighties continued through the Nineties as a result of steady improvement in care and the increasing expertise of the staff at the now well-established CF Centres. During the decade there were a number of notable additions to the treatments available in addition to further validation of existing treatments. These included rhDNase (Pulmozyme), a new preparation of tobramycin for inhalation (TOBI) and azithromycin. However, the provision of “optimal care”, which is complex and very expensive, for all people with CF, particularly in the UK, proved increasingly difficult, not least due its very high cost (Littlewood JM, Cross E. Present day treatment of cystic fibrosis: its content and cost. In: Clinical Economics in Gastroenterology. Bodger K, et al. (Eds), Blackwell Science, Oxford 2000: 220-249). In the UK, inadequate funding remained one of the major factors preventing the provision of optimal care for all.
Cross-infection between people with CF – a new major problem.
In 1979 Pseudomonas (now Burkholderia) cepacia in people with CF was first reported in North America and thereafter reports of this new pathogen, with the potential to spread between patients and cause serious illness, occurred with increasing frequency both from North America (Isles et al, 1984 above; Thomassen et al, 1985 above) and later from the UK (Simmonds et al, 1990 below). At first some clinicians were slow to accept that cross infection with B. cepacia between patients occurred until the early Nineties when this was established beyond any doubt (LiPuma et al, 1990 below; Govan et al, 1993 below). The severity and fatal nature of the associated illness (“cepacia syndrome”) in a proportion of infected patients with CF and its propensity to spread between patients has lead to a radical change in both clinic practice and the social habits of people with cystic fibrosis. Holiday camps for people with CF – so popular in N. America and so appreciated by some patients from the UK – were a source of cross infection and eventually abandoned (Pegues et al, 1994 below). In 1993, the Infection Control Group of the UK CF Trust recommended strict segregation of patients infected with Burkholderia cepacia.
This was the start of the era of cross-infection control, which was to radically alter the whole attitude to infection, cross-infection and social contact in CF Centres and in the community. Also there was increasing evidence of cross-infection with Pseudomonas aeruginosa. The concept of cross-infection between people with CF with organisms other than B. cepacia had not been regarded as a major consideration by the staff in most CF centres; the exception was the Danish CF Centre in Copenhagen where Niels Hoiby and his colleagues realised segregation of patients according to their microbiological status was important. Cross-infection with so-called ‘highly transmissible’ strains of Pseudomonas aeruginosa has been increasingly reported (Cheng et al, 1996 below; Jones et al, 2001 below) and there followed many reports of cross-infection with P. aeruginosa in cystic fibrosis. As a result of these developments it is was recommended that people with CF should be segregated according to their microbiological status and those with B. cepacia should also be segregated even from each other as those with less virulent genomovars (strains) may be infected by those with the more severe ones.
Other advances in treatment in the Nineties
Support for the routine use of prophylactic anti-staphylococcal antibiotics (flucloxacillin) in young children came from a controlled trial in screened CF infants in East Anglia in 1994 (Weaver et al, 1994 below), although some clinicians still considered the treatment predisposed to Pseudomonas aeruginosa infection.
Intravenous antibiotics were increasingly used for the chest infection in less severely affected patients at a much earlier stage rather than as a last resort – a principle central to modern treatment (“a matter of degree rather than kind”).
Early eradication treatment for early Pseudomonas infection became increasingly used in many European CF Centres.
Recombinant human DNase (Pulmozyme) was the first really effective mucolytic, which was very effective in reducing the viscosity of the sputum and improving respiratory function in a significant proportion of people with cystic fibrosis (Shak et al, 1990 below; Fuchs et al, 1994 below). It became an important and effective part of the daily treatment for the majority of patients with chronic infection and there was also evidence that even mildly affected patients may benefit (Harms et al, 1998).
A special preparation of tobramycin for inhalation (TOBI) became available in the Nineties. Although inhaled aminoglycosides (the IV preparations of gentamicin and tobramycin) had been widely used in the UK since Margaret Hodson’s 1981 paper (above), an important, well-conducted controlled trial to confirm the beneficial effect of this special preparation of tobramycin for inhalation (TOBI) was welcome (Ramsey et al, 1999 below). This preparation is now increasingly used.
From the early Nineties the new high strength pancreatic enzymes (Pancrease HL, Creon 25,000) were available and welcomed by people with CF many of whom, when taking a normal fat intake, did require a large number of standard enzyme capsules. So it was not surprising that patients, and their professional advisers, welcomed the introduction of the new “high lipase enzymes” in 1992, However, in 1993 a new unexpected and serious complication, fibrosing colonopathy with colonic strictures, was observed in Liverpool and eventually related to the very high doses of enzymes some patients were taking (Smyth et al, 1994 below); subsequently further patients with colonic strictures were reported from UK and the United States. Studies in the UK (Smyth et al, 1995 below) and the US (FitzSimmons et al, 1997 below) showed a relationship with the very high doses of lipase achieved with the new enzymes but the studies differed on showing an association with the copolymer covering of some preparations – the UK study suggested this was a factor. The problem has since receded in the UK following advice from the Committee on the Safety of Medicines to restrict the daily dose to an equivalent of lipase to not more than 10,000 IU /kg/day (Littlewood, 1999 below).
With increasing survival, problems related to CF including diabetes mellitus, liver disease, osteoporosis, pregnancy and fertility became increasingly common and presented further major management problems. New challenges, beyond imagination of the early CF clinicians, include the management of pregnancy in women with CF and successful treatment of infertility in men with cystic fibrosis. Also it is encouraging that even the problems of old age and CF are now being addressed.

Fig 1. Rosie Barnes OBE
In 1996 Mrs Rosie Barnes (fig. 1) was appointed as Chief Executive of the UK Cystic Fibrosis Trust. Rosie, as she is affectionately known, was a Member of Parliament (SDP) for Greenwich between 1987 and 1992. Prior to that she was involved in market research and marketing. Her interests in Parliament included health, education and the problems of children in care. She joined the Cystic Fibrosis Trust in 1996, after four years as Director of WellBeing, formerly Birthright.
Rosie has had a major impact on the work, development and influence of the CF Trust. In addition to the annual income increasing year by year from £3 million to £11 million over the next decade, it was under her initiative that the three leading gene therapy research groups in the UK came together to form the collaborative Gene Therapy Consortium in an endeavour to speed the translation of the gene discovery into the treatment of patients.
This it was now regarded as an important major decision focusing the major part of the CF Trust’s research funds on treatment of the basic defect by gene therapy. Also the CF Trust, with the advice of a specially appointed Scientific Advisory Committee, guaranteed ongoing financial support for the Gene Therapy Consortium – eventually amounting to £30 million during the decade.
Unfortunately as the financial recession deepened in 2010/11 the CF Trust was unable to provide the promised funding for the clinical trial towards which the Gene Therapy Consortium had been working for 10 years. Fortunately the Consortium were successful in achieving full funding (£3.1 million) for the trial from the Medical Research Council and the National Institute for Health Research through the Efficiency and Mechanism Evaluation Programme and the gene therapy trial started in 2012/13 and finished in 2014.
The results were published in 2015 (https://pubmed.ncbi.nlm.nih.gov/26149841/.) Full article on Pubmed the summary as follows –Monthly application of the pGM169/GL67A gene therapy formulation was associated with a significant, albeit modest, benefit in FEV1 compared with placebo at 1 year, indicating a stabilisation of lung function in the treatment group. Further improvements in efficacy and consistency of response to the current formulation are needed before gene therapy is suitable for clinical care; however, our findings should also encourage the rapid introduction of more potent gene transfer vectors into early phase trials.
The formation of the Gene Therapy Consortium and the provision of some £30 million by the CF Trust over ten years were obviously central to facilitate the freedom of the researchers to pursue various avenues of research to a stage where the members of the GTC could apply successfully to the MRC and NIHR for major on going financial support for their clinical trail; their success being a measure of the excellence of their work.
As I review this introduction to the Nineties in 2023 it is disappointing that there is still no effective gene therapy available for people with CF. Also, the dramatic effect of the new modulators, particularly Trikafta / Kaftrio, over the past decade has somewhat overshadowed the area of gene replacement. However, active research has continued as gene therapy is required for the 10% of people with CF who are not eligible for modulator treatment. It is encouraging that for these patients there is increasing interest and activity in the area of gene replacement therapy in a number of major centres including the CF Foundation. .
Clinical references in chronological order
1990 Pasque MK, Cooper JD, Kaiser LR, Haydock DA, Triantafillou A, Trulock EP. Improved technique for bilateral lung transplantation: rationale and initial clinical experience. Ann Thorac Surg 1990; 49:785-791. [PubMed]

Fig 2. Michael Pasque
The first bilateral lung transplantation in CF was performed in Toronto in 1988 and 17 were carried out between 1988 and 1991 (Ramirez JC et al. J Thorac Cardiovasc Surg 1992; 103:287-293. [PubMed] This improved operation was done through a transverse thoracosternotomy and involves sequential replacement of the two lungs. Positive features included separate bronchial anastomoses to reduce ischemic airway complications and elimination of the need for total cardiopulmonary bypass. Three patients were reported, one had CF, all recovered without complications and post operative function was excellent.
The double lung transplant operation gradually replaced heart-lung transplants for people with cystic fibrosis. Professor Michael Pasque (fig. 2) is a cardiothoracic surgeon and Professor of Surgery in the University of Washington School of Medicine, St Louis.
1990 Knowles MR, Church NL, Waltner WE, Yankaskas JR, Gilligan P, King M, Helms RW, Boucher RC. A pilot study of aerosolized amiloride for the treatment of lung disease in cystic fibrosis. N Eng J Med 1990; 322:1189-1194.[PubMed]
Michael Knowles (Fig. 2a) first discovered the increased bioelectrical potential difference across respiratory epithelium in CF (Knowles M et al. N Eng J Med 1981; 305:1489-1495 above).

Fig.2a Michael Knowles. Author’s photo
In this present study Knowles and Boucher investigate whether the inhibition of excessive absorption of sodium by inhaled amiloride might favourably affect the course of CF lung disease. Fourteen of 18 patients completed a one year double-blind, crossover trial comparing aerosolized amiloride (5 mmol per litre; 3.5 ml four times daily) with control solution. The mean (+/- SEM) loss of forced vital capacity (FVC) was reduced from 3.39 (+/- 1.13 ml) per day during treatment with vehicle alone to 1.44 (+/- 0.67) ml per day with Fig. amiloride (P <0.04). Sputum viscosity and elasticity, mucociliary and cough clearance improved during treatment with amiloride but the effect of amiloride was modest and short-lived; also the loss of FVC in the control group seemed excessive. Apparently the effect of inhaled amiloride is very transient and subsequently longer acting analogues were explored.
However, this report did encourage us in Leeds to look at the effect of giving nebulised amiloride during the intravenous antibiotic treatment of exacerbations of respiratory infection in CF. We observed a definite but insignificant improvement in the early response to intravenous antibiotics in the amiloride group (Bowler et al, 1995 below). No benefit was seen from amiloride by Graham A et al (No added benefit from nebulised amiloride in patients with cystic fibrosis. Eur Respir J 1993; 6:1243-1248.[PubMed]) nor in a French multi-centre randomized double blind placebo controlled trial in patients more than 5 years old (Pons G, et al, Pediatr Pulmonol 2000; 30:25-31. [PubMed]
However, earlier Kohler et al showed inhaled amiloride improved mucociliary clearance in patients with CF (Kohler et al, Eur J Respir Dis 1986; 69 (Suppl 146:319-326); also the same group confirmed this with a larger study (App AM et al. Am Rev Respir Dis 1990; 141:605-612. [PubMed] Also Lindemann et al from Giessen had reported 50.4% more sputum was produced by autogenic drainage after inhalation of amiloride than after isotonic saline also visible liquefaction of secretions was noted by the physiotherapist and patients (Lindemann H et al. Elimination of secretions in CF patients under amiloride inhalation. Pneumologie 1990; 44:1148-1150 [PubMed] [German]).
Later more active and longer acting drugs (P- 680 & P- 522-O2-Parion Sciences/Gilead) that inhibit excess Na absorption showed more promise.
1990 Yacoub M, Banner NR, Khaghani A, Fitzgerald M, Madden B, Tsang V, Smyth R, Hodson ME. Heart lung transplantation for CF and subsequent domino cardiac transplantation. J Heart Transplantation 1990; 9:459-67. [PubMed]

Fig 3. Magdi Yacoub
Between September 1984 and October 1988, 27 patients underwent combined heart-lung transplantation for treatment of end-stage respiratory disease caused by CF: survival was 78% at 1 year and 72% at 2 years. Lung function was greatly improved after transplantation, and long-term survivors achieved an excellent quality of life. Lymphoproliferative disorders developed in two patients; these disorders regressed after a reduction in immunosuppression. Two patients required retransplantation: one because of obliterative bronchiolitis and the other because of recurrent respiratory infections associated with a moderate tracheal stenosis and severe deterioration in lung function.
A modification of the technique used for heart-lung transplantation allowed 20 hearts from cystic fibrosis patients to be used for subsequent heart transplantation for non-CF patients.
This report is by Mr Yacoub (Fig.3) and his team, who performed the first heart lung transplantations in people with CF at Harefields Hospital, London in 1984. There were also reports from Mr Wallwork’s unit at Papworth, Cambridge (Penketh et al, 1987 above; Jones et al, 1988 above) and later from Professors Dark and Paul Corris from Newcastle.
1990 Colombo C, Battezzati PM, Crosignani A, Assaisso M, Ronchi M, Giunta A. Effects of taurine and ursodeoxycholic acid on liver function tests in patients with cystic fibrosis. Acta Universitatis Carolinae – Medica 1990; 36:148-151.[PubMed]
This was a memorable presentation by Professor Carla Colombo (fig. 3) of Milan at the European CF Society meeting in Prague in 1989 which I was fortunate to hear. It was the first report of the beneficial effect of oral ursodeoxycholic acid (URSO) in people with CF associated liver disease – before this there was no treatment for the liver disease. In nine CF patients with clinical and biochemical evidence of liver disease, taurine (30 mg/kg/day) was administered one month before and during the successive treatment with ursodeoxycholic acid (10-15 mg/kg/day). Standard liver function tests were determined before and after each period of treatment. Taurine administration produced only inconsistent changes of liver function tests from baseline, whereas after the addition of ursodeoxycholic acid there was a substantial improvement in all abnormal liver function tests (details reported in J Pediatr 1990; 117:482-489 below).
1990 Colombo C, Setchell KD, Podda M, Crosignani A, Roda A, Curcio L, Ronchi M, Giunta A. Effects of ursodeoxycholic acid therapy for liver disease associated with cystic fibrosis. J Pediatr 1990; 117:482- 489. [PubMed]

Fig 3. Carla Colombo. Author’s photo
The published report from Milan of data presented in 1989 at the European CF Society Meeting in Prague (1990 above) reporting the favourable effect of ursodeoxycholic acid (URSO) treatment 10-15 mg /kg day in 9 patients with CF who had chronic liver disease (presumably the same patients as reported in Prague in 1989). Liver function tests improved significantly (AST- by 34%, alanine aminotransferase – by 41%, gamma gutamyl transpeptidase – by 41% and alkaline phosphatase– by 19%).
Subsequently URSO was widely used in people with CF associated liver disease and appeared to represent one of the major advances in CF management. A further trial was published in 1996 (Colombo et al, 1996). Much information was published supporting URSO treatment at an early stage of liver involvement and eventually most people with CF who had evidence of liver involvement were treated with URSO.
However, a Cochrane Review last updated in 2017 concluded “There are few trials assessing the effectiveness of ursodeoxycholic acid. The quality of the evidence identified ranged from low to very low. There is currently insufficient evidence to justify its routine use in cystic fibrosis”.
In this writer’s opinion this conclusion was unfortunate as it could influence some clinicians, to withhold treatment from people with liver involvement who may have benefitted. The following statement would have been more appropriate – “There is a considerable amount of evidence that URSO improves liver function in people with CF particularly if used at an early stage; but, as yet, there is no clinical trial that conforms to Cochrane standards to confirm this”.
1990 LiPuma JJ, Dasen SE, Nielson DW, Stern RC, Stull TL. Person-to-person transmission of Pseudomonas cepacia between patients with cystic fibrosis. Lancet 1990; 336(8723):1094-6. [PubMed]

Fig 4 John LiPuma
Ribotyping, a method of strain identification based on analysis of bacterial genomic restriction fragment length polymorphisms, was used to investigate the acquisition of Pseudomonas cepacia by a patient with cystic fibrosis. Analysis of isolates recovered from the index patient and his contacts showed person-to-person transmission of this opportunist organism.
This documentation of the transmission of P. cepacia from one cystic fibrosis patient to another suggests that measures to limit the acquisition of the pathogen by patients with cystic fibrosis may be worth while.
Dr J J LiPuma (fig. 4) is Professor of Epidemiology and Pediatrics in the University of Michigan.
– This was the first clear report that there was significant person-to-person transmission but it was not until 1993 that person to person transmission was generally accepted in the UK following John Govan’s 1993 paper (Govan JR et al, 1993.[PubMed]) after which the CF Trust issued firm guidelines on the precautions necessary to prevent cross infection.
1990 Simmonds EJ, Conway SP, Ghoneim ATM, Ross H, Littlewood JM. Pseudomonas cepacia: a new pathogen in patients with cystic fibrosis referred to a large centre in the United Kingdom. Arch Dis Child 1990; 65:874-877. [PubMed]

Fig 4a. Eddie Simmonds. Authors’ photo
Although the first reports from N. America of this organism appeared in the late Seventies (Lararya-Cuassay et al, 1977 above), this report from Leeds was the first to report Burkholderia cepacia in the UK – previously known as Pseudomonas cepacia. We were impressed by the very serious consequences of this infection in some patients. Some clinicians in the UK still doubted the serious nature of this infection although three of our 11 patients died – two after an alarming and rapid deterioration now described as the “cepacia syndrome”. We could not identify any source of cross infection in the Leeds CF centre at that stage nor was there an inappropriate use of antibiotics. However, in retrospect, we suspected that at least two of our patients had acquired the infection at CF holiday camps in N. America some months before the organism appeared in their respiratory cultures.
Dr Eddie Simmonds (fig. 4a) was CF Research Fellow in Leeds and subsequently appointed as Consultant Paediatrician in Coventry where he developed the CF service.
A further report of 13 infected patients (three of whom died) from the paediatric CF centre in Manchester in the UK also failed to show evidence of cross infection and the authors suggested that further studies were required before segregation of patients should be recommended (Gladman G et al, Arch Dis Child 1992; 67:192-195.[PubMed].
It was not until 1993, following Professor Govan’s publication from Edinburgh (Govan et al, 1993 [PubMed]), that the UK CF Trust’s advisory group recommended strict segregation of all B. cepacia infected patients.
1990 Strandvik B, Hollsing A, Mollby R, Granstrom M. Antistaphyloccocal antibiotics in cystic fibrosis. Infection 1990; 18:48-50. [PubMed]

Fig. 4b Birgitta Strandvik
Chronic Staphylococcus aureus infection was present in 40-50% of the Stockholm patients. The presence of IgG ELISA serum anti-Staphylococcal antibodies reflected the presence and severity of the infection. The authors suggested the raised antibodies may indicate significant tissue damage and more severe disease and would be an indication for treatment.
This paper supported the use of an aggressive anti-Staphylococcal policy originally advocated by David Lawson, both involving the long term use of an antistaphyloccocal antibiotics with additional antibiotics when required.
In our Leeds clinic such a policy resulted in a low overall chronic S. aureus infection rate – in the whole clinic of only 14.5% with no children chronically infected below the age of 5 years and of only 8.3% of those less than 10 years old infected (Southern KW et al.1993 ECFS Madrid); also some of these children had been referred to the clinic when already chronically infected.
Later the clinical trial of Weaver et al, (1994 below) supported the use of long term flucloxacillin therapy up to the age of two years of age. Birgitta Strandvik later noted that despite long term flucloxacillin treatment it was rare to isolate methicillin resistant strains (Strandvik B. Ann Nestlé (Engl) 2006; 64:131-140); this was also our experience in Leeds – MRSA appeared to be acquired from others with that infection rather than developing as a result of receiving longterm flucloxacillin.
It was interesting that we noticed the few children who did grew S. aureus despite apparently taking flucloxacillin, also had low vitamin E levels and when their routine clinic urine specimen was checked for antibiotic activity there was none!! Adherence, or lack of it, seemed to be a definite possibility!!
1990 Shak S, Capon DJ, Hellmiss R, Marsters SA, Baker CL. Recombinant human DNase 1 reduces the viscosity of cystic fibrosis sputum. Proc Natl Acad Sci 1990; 87:9188-9192.[PubMed]

Fig 5. Jim Littlewood , Steve Shak and Steve Conway in 1995. Author’s photo.
An early report of the significant effect of recombinant human DNase1 (Pulmozyme) on sputum viscosity in people with CF by Steve Shak (Fig. 5). To evaluate the potential clinical utility of recombinant human DNase I (rhDNase) in the treatment of CF, the authors cloned, sequenced, and expressed rhDNase. Catalytic amounts of rhDNase greatly reduced the viscosity of purulent CF sputum, transforming it within minutes from a non-flowing viscous gel to a flowing liquid. The reduction in viscosity is associated with a decrease in size of the DNA in the sputum.
The authors suggested that inhalation of an rhDNase aerosol may be a simple direct approach that would help individuals with CF, and other patients with pneumonia or bronchitis, to clear their airways of purulent secretions. The definitive Phase III trial was published in 1994 – Fuchs HJ et al. Effect of aerosolized recombinant human DNase on exacerbations of respiratory symptoms and on pulmonary function in patients with cystic fibrosis. The Pulmozyme Study Group. N Eng J Med 1994; 331:637-642. below).
Pulmozyme was undoubtedly one of the major clinical advances of the decade and was licensed for people with CF by the FDA in 1994. Regular treatment with DNase would become widely used (by 78.4% of people with CF over 6 years old on the CFF registry in 2010) first amongst those with significant chest involvement and then later for those with milder chest problems (Quann et al, 2001 below) and eventually, in some CF units, for infants. The cost (approximately £7000 per year) proved a problem in certain parts of the UK.
As far back as 1950 Armstrong JB & White JC (Lancet 1950; 2:739-742. [PubMed]) had shown that bovine pancreatic DNase1, added to viscid purulent sputum, destroyed the extra cellular fibres of DNA and reduced the viscosity. Later Elmes PC & Armstrong JB (Thorax 1953; 8:295-300. [PubMed]) reported its use in chronic bronchitis, but the side effects of the bovine preparation eventually precluded its use and further development at that time (Raskin P. Am Rev Respir Dis 1968; 98:697-698. [PubMed]). Subsequently the rhDNase (Pulmozyme) reported here proved to be one of, if not the, major therapeutic advance of the Nineties. Numerous subsequent reports confirmed both short and long term benefits.
1990 Regelmann WE, Elliot GR, Warwick WI, Clawson CC. Reduction of sputum Pseudomonas aeruginosa density by antibiotics improves lung function in cystic fibrosis more than do bronchodilators and physiotherapy alone. Am Rev Respir Dis 1990; 141:914-921. [PubMed]

Fig 6. Warren Regelmann
This widely quoted paper provided definite objective evidence of the beneficial effect of antibiotics over and above the other measures used when patients with chronic Pseudomonas aeruginosa infection were treated for a pulmonary exacerbation. The favourable effect of antibiotics had been questioned in an extraordinary publicationl from Toronto where it was stated “there was no difference in the course during the 6 to 24 months after the study period. Intravenous antibiotics are not essential in the management of all acute respiratory exacerbations of mild to moderate severity in patients with cystic fibrosis” (Gold R et al. J Pediatr 1987; 111:907-913. [PubMed] This was a finding quite out of keeping with the experience of most CF clinicians who, fortunately, did not heed the Toronto advice!
This present study has a rather complex design but as it is generally accepted as providing evidence of the beneficial effect of intravenous antibiotics so it is described here in some detail.
For the first 4 days of study, all patients received bronchodilating aerosols and chest physiotherapy but no antibiotics. During this time, the patients showed significant improvement in mean FVC, FEV1, and maximal midexpiratory flow rate (FEF25-75). In 12 of 13 trials, the patients showed no significant increases in the density of Pseudomonas aeruginosa during these first four days. In these 12 trials, the patients were stratified by their initial FVC and randomized to receive either parenteral tobramycin and ticarcillin (n = 7) or placebo (n = 5), and in addition to continuing aerosol and chest physiotherapy. In the remaining trial, the patient had a significant rise in the density of P. aeruginosa during the run in and therefore was assigned to the antibiotic group.
During the next 14 days of therapy, the antibiotic group showed significantly (p < 0.01) greater reductions in log10 colony-forming units (cfu) of P. aeruginosa per gram of sputum and greater increases in FVC, FEV1, and FEF25-75 than did the placebo group. The degree of decrease in log10 cfu P. aeruginosa/g sputum correlated significantly (p < 0.001) with the degree of improvement in FVC, FEV1, and FEF25-75.
Dr Warren Regelmann (fig. 6) is now an Associate Professor of Pediatrics, in the Divisions of Pediatric Pulmonary and Critical Care Medicine and Pediatric Infectious Diseases, University of Minnesota.
1990 Waters DL, Dorney SF, Gaskin KJ, Gruca MA, O’Halloran M, Wilcken B. Pancreatic function in infants identified as having cystic fibrosis in a neonatal screening program. N Eng J Med 1990; 322:303-308.[PubMed]

Fig. 6a. Donna Waters. sydney.edu.au
Assessment of pancreatic function in 78 children identified in the New South Wales neonatal screening program as having cystic fibrosis. Measurements of faecal fat excretion, pancreatic-stimulation tests, and estimations of the serum level of pancreatic isoamylase indicated that 29 of the 78 children (37 percent) had substantial preservation of pancreatic function. These 29 children (median age, four years) had growth that was close to normal and comparable to growth in children with severe pancreatic insufficiency who received oral enzyme therapy. Pancreatic insufficiency subsequently developed in 6 of the 29 patients, at 3 to 36 months of age. The authors concluded that the serum immunoreactive-trypsin assay used in neonatal screening programs identifies patients with CF who have sufficient pancreatic function to achieve normal fat absorption and that a substantial proportion of infants identified as having cystic fibrosis are in this category (also Gaskin et al, 1991 below)
Thirty four of these 76 children had their pancreatic function assessed an average of 2.3 years after diagnosis – so subsequently a further 20 infants were studied at the time of diagnosis (Gaskin et al, 1991 below).
The number of children who were pancreatic sufficient in this series is higher than expected but would depend on the proportion carrying a “mild” mutation. This paper was published in February 1990 and presumably submitted some months before that i.e. before the CF gene was identified in 1989.
Donna Waters (Fig.6a) eventually became Deputy Executive Dean and Professor in the Faculty of Medicine and Health University of Sydney. Her research interests include paediatrics and child health, evidence implementation (including innovations and strategies that aid the implementation of evidence into clinical practice), evaluation research, and research education.
1990 Walters MP, Kelleher J, Gilbert J, Littlewood JM. Clinical monitoring of steatorrhoea in cystic fibrosis. Arch Dis Child 1990; 65:99-102. [PubMed]
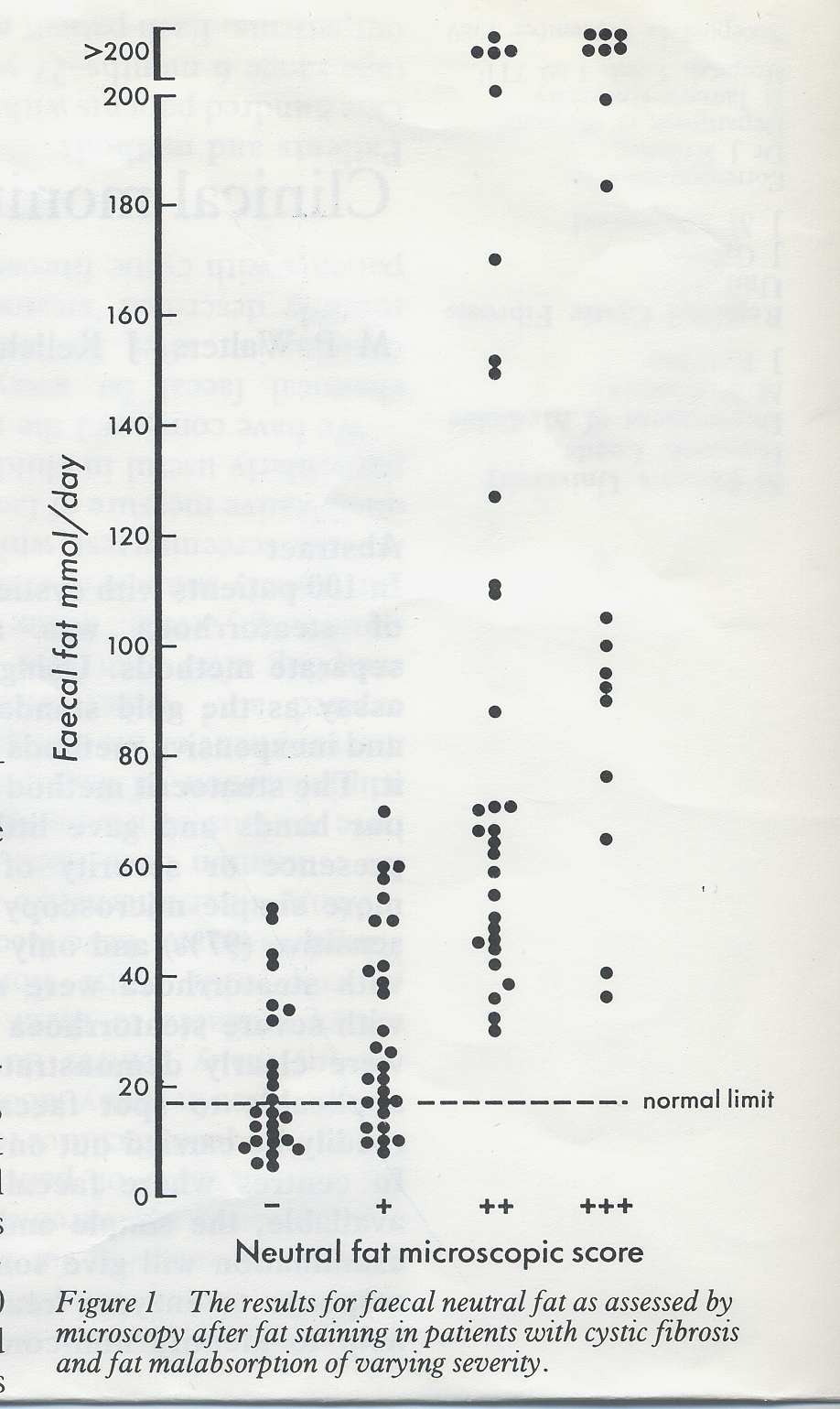
Fig 7. Faecal neutral fat – microscopy vs. chemical analysis.Walters et al, 1990
When compared with chemical faecal fat assays and steatocrit the simple microcopy method was highly sensitive (97%) and
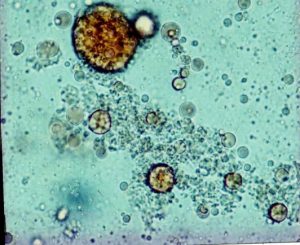
Fig 8. Faecal microscopy – fat globules. Walters et al, 1990.
only three of 80 patients with steatorrhoea would have been missed using this technique. All patient with severe steatorrhoea (> 60 mmol fat/day) were clearly identified (figures 7 & 8).
This method was used until recently in the Leeds CF Centre but due to financial cuts was withdrawn by the laboratory. It does give some indication as to the success of the enzyme replacement therapy as chemical estimations of faecal fat are rarely done unless part of a research project. It is of some concern that few clinics in the UK make any regular assessment of the severity of steatorrhoea which is unfortunate as severe fat malabsorption may be present without significant abdominal symptoms. Conversely severe abdominal symptoms may be present without there being steatorrhoea – a situation which obviously would not respond to increasing the enzyme dose as, unfortunately, must occur frequently in view of the unnecessarily large doses of enzyme taken by a significant number of people with CF in the UK (Mehta A. Further comments on the fibrosing colonopathy study. Lancet 2001; 358:1547-1548).
1990 Kennedy JD, Dinwiddie R, Daman-Willems C, Dillon MJ, Matthews DJ. Pseudo-Bartter’s syndrome in cystic fibrosis. Arch Dis Child `1990; 65:786-787. [PubMed]
A further seven cases of Pseudo-Bartter’s syndrome are described from Great Ormond Street, London. Chronic salt depletion was associated with severe failure to thrive which was soon reversed when the salt deficit was corrected.

Fig 9. Robert(Bob) Dinwiddie and George Russell receive awards at the BPRS dinner in 2009
Dr Bob Dinwiddie (Fig. 9), the senior author, was consultant Respiratory Paediatrician at the Hospital for Sick Children, Great Ormond Street in London where he succeeded Dr Archie Norman as Director of the CF Unit. He was heavily involved in CF care and respiratory paediatrics both nationally and internationally until his retirement in the 2005.
1991 Gaskin K, Waters D, Dorney S, Gruca M, O’Halloran M, Wilcken B. Assessment of pancreatic function in screened infants with cystic fibrosis. Pediatr Pulmonol 1991; Suppl 7:69-71. [PubMed]
Previously these authors reported that 37% of infants with CF diagnosed by neonatal screening with the dried blood spot immunoreactive trypsin assay were pancreatic sufficient (Waters et al, 1990 above). However, 34 of the 78 infants had pancreatic function tests an average 2.3 years after diagnosis, thus it was possible that the percentage with neonatal pancreatic sufficiency was even underestimated, due to the loss of pancreatic function with time in some infants. To assess this hypothesis the authors assessed pancreatic function at the time of diagnosis in a further 20 infants since the completion of the previous study. Results of faecal fat determinations and/or pancreatic stimulation tests indicated that no less than 10 (50%) of these infants have pancreatic sufficiency.
Combining these results with those of the previous study, 31 of 64 patients (48%) have pancreatic sufficiency at this early age. The authors monitored the progression of pancreatic disease in the 39 children with pancreatic sufficiency recognized to date. Eleven have developed pancreatic insufficiency and require enzyme replacement therapy. Five others have shown further improvement of colipase secretion with age. So the authors confirmed their previous conclusion that the dried blood immunoreactive trypsin screening program for cystic fibrosis does recognize patients with pancreatic sufficiency and at diagnosis nearly half their patients were in this category. To date, 28% of patients with pancreatic sufficiency have demonstrated a variable decline in pancreatic function with age.
In this study there were a surprisingly large number of infants who were pancreatic sufficient (48%) and this is quite different from our experience of screened infants with CF over the last 30 years in Leeds, although, of course, the frequency will depend on the mutations which the infants have. For example of a recent 15 screened infants with CF in Leeds only 2 (13%) were pancreatic sufficient (Wolfe et al. J Cyst Fibros 2005; 4(S1):S94).
The faecal pancreatic elastase is now a convenient and reliable way of determining pancreatic function and following the progress of pancreatic function in these infants.
The lesson here is that not all newborns with CF require enzyme replacement therapy so it is important to make sure there is evidence of pancreatic insufficiency preferably before or soon after commencing enzymes – in practice now easily done with a faecal elastase measurement even if enzymes have been started to avoid delay. Also, if available, a small specimen of stool is saved for fat microscopy (Walters et al, 1990 above). The faecal elastase test is a reliable indicator of pancreatic function even when the infant is taking enzyme replacement therapy – in this respect it differs from faecal trypsin and chymotrypsin. This is very useful particularly when patients are referred to the CF Centre from elsewhere and already taking pancreatic enzyme supplements some having been started before malabsorption had been established.
1991 McElvaney NG, Hubbard RC, Birrer P, Chernick MS, Caplan DB, Frank MM, Crystal RG. Aerosol alpha-1-antitrypsin treatment for cystic fibrosis. Lancet 1991; 337:392-394.[PubMed]

Fig 10. Gerry McElvaney
A1-antitrypsin, the main inhibitor of neutrophil elastase, was given in aerosol form to 12 CF patients and found to suppress neutrophil elastase in respiratory lining fluid and restore its anti-neutrophil elastase capacity. Also, the treatment reduced the reversed inhibitory effect of CF epithelial lining fluid on Pseudomonas killing.
Apparently the material used in this trial (purified human plasma a1-antitrypsin – Prolastin, Cutter Biological) was very difficult to obtain in sufficient quantities.
A subsequent trial, with a genetically engineered product which eventually became available, disappointingly failed to show significant benefit to patients and was not further developed as a treatment for cystic fibrosis (Martin SL, et al, 2006 below). However, some interest continues in Germany and there may be further developments (Griese M et al. alpha1-antitrypsin inhalation reducues airway inflammation in cystic fibrosis patients. Eur Respir J 2007; 29:240-250. [PubMed] Free article).
Prof Noel G McElvaney (fig 10) is Head of the Department of Medicine and School of Medicine, RCSI.
1991 Laroche D, Travert G. Abnormal frequency of delta F508 mutation in neonatal transitory hypertrypsinaemia. Lancet 1991; 337:55. [PubMed]
Neonatal CF screening programmes that involve DNA testing for CF mutations identify some infants who are CF carriers. There was much discussion as to whether parents should be told their infant is a carrier of one CF mutation although most clinicians, including the present writer, were quite firmly in favour of informing the parents. Most generally agreed that it was acceptable to detect CF carriers provided adequate genetic advice was provided to the parents and child bearing relatives for planning future pregnancies now it was known that one of them may be a CF carrier.
1991 Malfroot A, Dab I. New insights on gastro-oesophageal reflux in cystic fibrosis by longitudinal follow up. Arch Dis Child 1991; 66:1339-1345. [PubMed]
Gastro oesophageal (GO) reflux was first described as a problem in CF by Jean Feigelson (Feigelson et al, 1975 above) and subsequently the problem was thought to be related to progressive CF in older children. In this Belgian study 21 of 26 (81%) young children with CF aged less than 60 months were studied and 20 confirmed to have reflux by oesophageal pH tracings. Sixteen improved with anti-reflux treatment with improved weight gain, less cough and wheeze but half still had the reflux one year later. The authors concluded the reflux was not caused by the CF chest problems as it improved with time – at the same time as the CF gets worse – hence their title “new insights into GO reflux”.

Fig. 11 Anne Malfroot
There was to be continuing interest in GO reflux both in respect to physiotherapy practices in CF infants (Button et al, 1997 below) and in adults where GO reflux was shown to be frequent and important in exacerbating respiratory symptoms (Scott RB et al, 1985; Ledson MJ et al, 1998) particularly in relation to patients after lung transplantation (Button BM et al. J Heart Lung Transplant 2005; 24:1522-1529). Newer techniques of oesophageal pH monitoring and also fibreoptic endoscopy allowed more frequent recognition and more accurate diagnosis.
Dr Anne Malfroot (fig. 11) is Professor in Pediatrics at the Vrije Universiteit Brussel VUB University in Brussels. She is the Head of the Clinic of Pediatric Respiratory Diseases, Infectious Diseases, Travel Clinic and Cystic Fibrosis Clinic at the Universitair Ziekenhuis UZ Brussel.
1991 Valerius NH, Koch C, Hoiby N. Prevention of chronic Pseudomonas aeruginosa infection in cystic fibrosis by early treatment. Lancet 1991; 338:725-726.[PubMed]

Fig 12. Henrik Valerius
A randomised controlled trial from Copenhagen confirming that early P. aeruginosa infection could be eradicated in 80% of patients with CF by three weeks treatment with oral ciprofloxacin and nebulised colistin. The infection became chronic in only 2 of 14 (14%) of treated patients but in 7 of 12 (58%) of the controls.
The authors comment – “Our results thus confirm and extend the preliminary report by Littlewood et al, colleagues (1995 above) who used colistin inhalations. Since chronic colonisation with Ps aeruginosa is associated with increased morbidity and mortality we recommend the use of anti-Pseudomonas treatment whenever Ps aeruginosa is isolated from the sputum of cystic fibrosis patients”
In 2012 Dr Valerius (fig. 12) kindly provided this image and also commented as follows – “Your comments on our paper are very flattering. It is the only paper on CF I ever published. My main interest having been infectious diseases and immunology, in piarticular neutrophil function. I had very close collaboration with Dr Christian Koch on this (he was in many ways my mentor, also long before he became involved in CF). I was involved in clinical work with CF in 3 periods of my professional life, from 1971-1973 as resident which was my first appointment in paediatrics and again through 1980 after returning from a research fellowship at Harvard Medical School. I wrote from Boston to Dr EW Flensborg and asked if he had a position when I would be back and he wrote back I should just say when, which was very generous indeed. Finally from 1985 until 1988, when I was appointed consultant and clinical lecturer at Department of Paediatrics at Copenhagen University Hospital at Hvidovre being mainly involved in paediatric HIV until I retired 2 years ago in 2010.
I presented the paper first as a poster at the international CF Conference in Washington in 2000. It is kind of ironic that I think this poster was by far the least studied by the participants at the conference. As I recall none at all stopped by to read or discuss it – the only reaction I remember was head shaking! No bitterness now whatsoever – it’s kind of fun now and it was then as I knew I had left CF forever. But I was very surprised when the Lancet accepted it – well you always get surprised when the Lancet or NEJM accept something!”
Finally Dr Valerius modestly observes that his main contribution to CF was probably not this paper but the fact that in 1973 his sister-in-law, Annelise Hansen (figure 12a), was appointed nurse at the CF out patient clinic where she stayed for more than 30 years becoming almost an institution there!
Fig. 12a. Annelise Hansen
Indeed I also remember Annelise very well and being very impressed by her when I visited the Copenhagen CF Centre for 2 days in 1997. Later describing my visit to the outpatient clinic I wrote “One is immediately conscious of a friendly and welcoming atmosphere and a good easy relationship between patients, parents and clinic staff. The senior clinic nurse, Annelise, has worked there for 28 years and combines the functions of clinic nurse manager, CF Nurse Specialist, and friend and adviser to the patients and their families” (A Visit to the Copenhagen CF Centre. Jim Littlewood. 1997. Pharmax Ltd). I recall asking Annelise if she had any problems dealing with both the children and the adults with CF. She replied that she and the staff had known the patients since they were small children and it was only natural that the relationship should continue when they became adults. Certainly the arrangement seemed to work very well in Copenhagen although recently, in view of the increasing number of adult patients, an adult CF unit opened there.
In the present paper, Dr Valerius and the authors generously refer to our short letter to the Lancet in 1985 which was the first report on the use of inhaled colomycin to eradicate early Pseudomonas infection in CF and apparently stimulated their study (Littlewood JM, Miller MG, Ghoneim AT, Ramsden CH. Nebulised colomycin for early Pseudomonas colonisation in cystic fibrosis. Lancet 1985; i: 865. [PubMed]). Although only a letter this was undoubtedly the most important publication of my career! It was good that the Copenhagen team confirmed our observations in their controlled trial. Prior to treating CF children with inhaled colomycin I had been impressed by a paper from the Sixties describing successful treatment of Pseudomonas aeruginosa respiratory infections in non-CF patients with a combination of IV and inhaled colistin (Halliday NP. Clinical Trials Journal. August 1967. 771-775) and also by an editorial by Wallace Herrell in Clinical Medicine (September 1968 18-19) of successful aerosolized treatment of 60 patients with gram negative pneumonia, 30 of whom were infected by P. aeruginosa, the majority of whom improved; P. aeruginosa was eliminated from their sputum. The editorial concluded – “Thus it would appear that sodium colistimethate can be administered safely as an aerosol and it appears to be quite effective in the treatment of pulmonary infections. including pneumonia, owing to sensitive organisms particulalry the Pseudomonas strains”.
After discussions with the firm (Pharmax Ltd – now Forest Laboratories) as to the dose, nebuliser etc in 1983/4 we started to treat children as soon as P. aeruginosa was cultured. Even though the numbers in the present Valerius et al 1991 trial were small and the patients not formally randomised, this was certainly one of the most important CF papers of the decade and confirmed that early Pseudomonas infection could be eradicated with nebulised colomycin and oral ciprofloxacin. It is difficult to understand why this trial from Copenhagen, which so clearly contradicted the previous widely held belief, that it was impossible to eradicate Pseudomonas once cultured, was not followed by the widespread introduction of early eradication treatment for P. aeruginosa. Fortunately a few centres in Europe did introduce early eradication treatment but they were a minority. The fact that early treatment of Pseudomonas was so slow to be introduced in the UK and much of Europe (with notable exceptions) and was still not recommended in the USA over a decade after this report, was difficult to explain.
It was predictable that these varied approaches to early treatment of Pseudomonas were reflected gradually in the markedly different prevalence of chronic Pseudomonas infection in different CF centres- this difference in the prevalence of chronic infection became increasingly obvious first in paediatric patients as time progressed (Frederiksen et al, 1996; Lee et al, 2003; Lebecque et al, 2006 – all below).
It is of interest that even in 1998, reviewing the history of Pseudomonas infection in people with CF, a highly regarded US CF centre director wrote – “early administration with aerosol colistin may delay colonisation with P. aeruginosa. This intriguing observation has not been verified by prospective controlled studies” (Ramsey BW. Pediatrics 1998; Supplement: 210-213) – even though by this time early eradication of P. aeruginosa was widespread practice in Europe and already supported by many publications in addition to that of Valerius et al, 1991 from Copenhagen (Brett MM et al. Arch Dis Child 1992; 67:1086-1088.[PubMed]; Frederiksen B et al, Pediatr Pulmonol 1997; 23:330-335.[PubMed]; Weisemann HG, et al. Pediatr Pulmonol 1998; 25:88-92. [PubMed]; and later Munck A et al. Pediatr Pulmonol 2001; 32:288-292). [PubMed]
Fig 13 Christian Koch
In 1997 I had the good fortune to interview the late Dr Christian Koch (figure 13), then the Medical Director of the Copenhagen CF centre, for the video. When I asked him at the end of the day what aspect of CF treatment he regarded as the most important, he thought for some time and then replied – “When I look back on what we’ve done all through the years that I’ve been involved with cystic fibrosis, I would say that the early treatment of Pseudomonas is probably the best thing that we have done for the patients. It becomes more and more clear that really what determines the long term course is whether you get Pseudomonas or not” .
1991 Hodson ME, Madden BP, Steven MH, Tsang VT, Yacoub MH. Non-invasive mechanical ventilation of CF patients – the bridge to transplantation. Eur Resp J 1991; 4:524-7.[PubMed]
This technique of non-invasive ventilation was developed as a direct result of attempting to prolong the survival of people with CF awaiting a heart-lung transplant at the Brompton Hospital – prior to the possibility of heart-lung transplantation assisted ventilation was usually regarded as inappropriate for people with CF in respiratory failure as their prognosis was uniformly bad. The case histories of six patients with CF using nasal ventilation while awaiting heart-lung transplantation are reviewed; four of the patients did well.
This method of ventilation proved to be a useful bridge to transplantation when a patient suddenly deteriorates. This is also a very cost effective method of ventilation and does not encroach on conventional Intensive Care Unit facilities.
1991 Morrison G, Morrison JM, Redmond AOR, Byers CA, McCormack KJ, Dodge JA, Guilford SA, Bowden MW. Comparison between a standard pancreatic supplement and a high enzyme preparation. Aliment Pharmacol Ther 1992; 6:549-555.[PubMed]
First of a number of reports showing new “high strength” enzymes were effective. This study compared the relative effectiveness of a standard pancreatic enzyme supplement (‘Creon’, Duphar) and a new high strength preparation (‘Pancrease HL’, Cilag) containing about 3 times the lipase and more than 5 times the protease activity. Capsule dosage was adjusted to a ratio of approximately 3 Creon to 1 Pancrease HL to provide similar intakes of lipase. Fat balances showed that absorption of fat did not change significantly on conversion to the new high-lipase product, and the coefficient of absorption of total energy was similarly maintained. The coefficient of protein absorption was significantly enhanced with the high enzyme preparation (P < 0.01), which may explain the reported subjective improvement in stool odour. No adverse effects were recorded. Patient acceptability of the new compound was high; the great reduction in the number of capsules required at each meal was cited by all patients as the reason for their preference.
Later reports associated fibrosing colonopathy with the use of Pancrease HL but not with the new high strength Creon 25,000 (Smyth et al, 1994. below) which some, including this writer, considered due to the presence of the copolymer, Eudragit, in the covering of all the new high strength enzymes other than Creon 25,000.
1991 Hammond KB, Abman SH, Sokol RJ, Accurso FJ. Efficacy of statewide neonatal screening for cystic fibrosis by assay of trypsinogen concentrations. N Eng J Med 1991; 325:769-774.[PubMed]

Fig 14. Keith Hammond
Keith Hammond (fig. 14) was one of the early enthusiasts for neonatal CF screening and was the biochemist involved with these screening studies from Denver, Colorado on 278,399 infants from 1982 to 1987, using immunoreactive trypsin. They confirmed that the method was feasible and could be implemented with acceptable rates of repeat testing and false positives and false negatives. 95% of infants with CF who did not have meconium ileus could be identified by the screening method.
Many subsequent valuable reports have resulted from this Denver screening programme including the early bacteriologic and clinical course (Abman SH et al. J Pediatr 1991; 119:211-217.[PubMed]), early respiratory course (Accurso FJ et al. Pediatr Pulmonol Suppl 1991; 7:42-45. [PubMed]), fat soluble vitamin status (Sokol RJ et al. Pediatr Pulmonol Suppl 1991; 7:52-55.[PubMed]) and pancreatic and nutritional state (Bronstein MN et al. J Pediatr 1992; 120:533-540.[PubMed]
1991 Scott-Jupp R, Lama M, Tanner MS. Prevalence of liver disease in cystic fibrosis. Arch Dis Child 1991; 66:698-701.[PubMed]

Fig.14a Robert Scott-Jupp
A search for the presence of liver disease among 524 patients with CF in the UK by Dr Robert Scott-Jupp (Fig.14a) working with Prof. Stuart Tanner in Leicester; details of a further 576 patients were obtained from databases. The overall prevalence of overt liver disease as indicated by the presence of an enlarged liver or spleen (or both) was 4.2%. The age related prevalence rose to a peak in adolescence, and then fell in patients over 20 years old. The implied increase in mortality among those with liver disease was not explained by deaths from liver disease, which were rare. Male patients were significantly more affected than female, the ratio being 3:1 among adolescents. Increasing prevalence of liver disease in patients with cystic fibrosis was not just a result of longevity.
1991 Warwick WJ, Hansen LG. The long-term effect of high frequency chest compression therapy on pulmonary complications of cystic fibrosis. Pediatr Pulmonol 1991; 11:265-271.[PubMed]

Fig 15. Warren Warwick. Author’s photo
A high-frequency chest compression (HFCC) device for clearance of mucous secretions from airways was tested in 16 patients with CF with significant improvement in pulmonary function for thC period, which averaged 22 months per patient. The device consists of a variable air pulse delivery system and a non-stretch inflatable vest worn by the patient to cover the entire torso. Ninety-four percent of patients line slopes for percent predicted forced vital capacity (FVC) and forced expiratory volume in 1 second (FEV1) became more positive during self-administered HFCC therapy as compared to slopes before HFCC therapy, when manual chest physical therapy was used. Although this device (figure 14) has never found favour amongst physical therapist in the UK, over the next 15 years “the vest” became popular and widely used in North America by some 60% of people with CF and by many others with chronic respiratory disorders.
The device has been pioneered by veteran CF centre director Warren Warwick (figure 15) from Minnesota and has been the subject of many subsequent publications showing comparable efficiency with other forms of physiotherapy and improvements with alterations in the wave form created. The first reports on high frequency chest wall compression were from King M et al.(Am Rev Respir Dis 1983; 128:511-515.[PubMed]; Am Rev Respir Dis 1984; 130:703-706.[PubMed]
In 2016 Warren Warwick (fig. 15) (1928-2016) died aged 88 years.
Preston Campbell III (CEO of the CF Fundation) wrote ” It is with a heavy heart that I share the passing of Warren J Warwick MD who died in Feb. 15th 2016. Dr Warwick had a profound and lasting impact on the CFF through his scientific expertise and his long standing commitment to his patients”

Fig. 15a Warren Warwick, John Dodge & Jean Fiegelson. Author’s photo
Professor John Dodge wrote – Warren was 88 years old when he died. He had many skills, and many achievements, but his outstanding quality was an unrivalled dedication to improving the quality and duration of the lives of CF patients. Data from his clinic in Minneapolis shows that his patients lived about a decade longer than those in other CF centres worldwide. To achieve this result he had developed or recruited expertise in epidemiology, statistical analysis, pulmonary physiology, immunology, infectious disease control, pathology, physiotherapy, gastroenterology, nutrition, endocrine and diabetic care, nursing, adult medicine, genetics, psychology, and engineering. He worked with pharmaceutical and medical device companies, and with charities and voluntary agencies. He built a motivated team, many members of which have become eminent in their own right inspired by his leadership and encouragement. The extra ingredient was patient and family contact time. Warren routinely spent 45 minutes with each consultation, checking medication doses and adherence to treatment, and empowering his patients to adapt their own regimes in (sometimes unorthodox) ways which would help them maintain or improve their clinical state. He helped develop the percussive vest for CF because human physiotherapists were not always available and were costly. He set no limits to life expectancy. He never promoted himself. He simply examined all the evidence and went wherever it took him. Although tenacious and sometimes iconoclastic, he was gently spoken and avoided confrontation. But the success of his clinic became public news when it was highlighted in an article in the New Yorker in 2004 which resulted in CF patients from all over the USA and beyond seeking a consultation. His Annalise Marzotto professorial chair in the University of Minnesota was named for an Italian patient who had died there.
In 2004 in an excellent article by Atul Gwande in The New Yorker, Warren Warwick’s method of working with patients was described – “The Bell Curve. What happens when patients find out how good their doctors really are?’ which can be highly recommended. The full text is available on the internet (Publications The New Yorker/Atul Gwande). Also suggest my 2004 entry in this present history for Atul Gwande.
I met with Warren many times at CF Conferences. He knew I had some involvement with The UK CF Trust and that I was involved in the early stages of the UK Database with the Dr Mehta and his wife Gita in Dundee. He was interested in our progress and always encouraging. It was always a pleasure to talk with him. Warren was truly one of the great pioneers of CF care – it was indeed a privilege to have known him.
1991 Chatfield S, Owen G, Ryley HC, Williams J, Alfaham M, Goodchild MC, Weller P. Neonatal screening for cystic fibrosis in Wales and the West Midlands: clinical assessment after five years of screening. Arch Dis Child 1991; 66:29-33.[PubMed] A UK neonatal CF screening study, funded by the Cystic Fibrosis Trust, using measurement of immunoreactive trypsin, was undertaken in Wales (Mary Goodchild) and the West Midlands (Peter Weller) on alternate weeks for five years from 1985. Fifty eight infants, not considered to be at risk of CF (i.e. they did not present with meconium ileus and did not have a sibling with cystic fibrosis) were detected by screening and were compared with 44 children born on the non-screening weeks whose CF was diagnosed clinically. The false negative rate in the screened group was a very disappointing 13.4%. Predictably, the screened group were diagnosed earlier and spent less time in hospital. In other respects the groups were similar to the age of 4 years.
Treatment of CF in 1985 in the UK was usually undertaken at the local hospitals (Littlewood et al, 1984 above, 1988 above and 1993 below) and half the children in this trial were only seen at their local hospitals by general paediatricians who had no particular expertise in CF – this appeared to be the major flaw of this Wales & West Midlands study. However, the study did show that if clinical care was not of a high standard there was no advantage in neonatal diagnosis. Subsequently this study was not considered to provide support for the introduction of national neonatal CF screening by the UK National Screening Committee.
There was a final report of this data from Cardiff by Iolo Doull (Doull IJ et al. Pediatr Pulmonol 2001; 31:363-366.[PubMed] Eligible children with CF who died in the first five years of life were identified from the local paediatricians and from the National UK CF Survey. In all, 230,076 infants had been screened and 234,510 unscreened. 176 children with CF were identified, of whom seven died in the first five years of life, three having presented with meconium ileus. Median age of diagnosis in the screened group was eight weeks (this would not be regarded as acceptable now but DNA testing was not available in addition to IRT until after 1990). On an intention to treat analysis, all four non-meconium ileus-related deaths occurred in the unscreened group; however, the clinical presentation of two of these infants led to their being diagnosed prior to eight weeks, i.e., earlier than would have been likely by screening as practised in the study. The authors concluded that newborn screening has the potential to decrease infant CF deaths, but if it is to be successful, identification and treatment must occur as soon as possible after birth.
Eventually neonatal CF screening was agreed by the UK government in 2001 after the Cystic Fibrosis Trust made personal representation to Ms Yvette Cooper the then Health Minister. As a result of the overwhelming pressure from the CF Trust and the whole CF professional and lay community rather than on the advice of the National Screening Committee that, even then, still considered the evidence for screening inadequate even after the Government recommendation! The 2001 Farrell paper, which showed a long term nutritional advantage in the screened infants (Farrell et al, 2001; 107:1-13 below), was important in finally convincing the government to introduce national neonatal CF screening.
The major lessons from all this being that, of course neonatal diagnosis is essential but must be followed by good CF care from the start to prevent irreversible malnutrition and chronic respiratory infection.

Fig. 17 Henry Ryley

Fig 16. Peter Weller
Dr Peter Weller (fig. 16) was Director of the CF Centre at the Birmingham Children’s Hospital from 1980 to 2007 and closely involved with this screening study.
Dr Henry Riley, (fig. 17) was the microbiologist from Cardiff, who was closely involved with this and many other studies on CF and also with the European CF Society for many years.
1991 Warner JO. Heart-lung transplantation: all the facts. Arch Dis Child 1991; 66:1013-1017.[PubMed]

Fig. 18 John Warner
Professor John Warner (fig. 18), then paediatrician at the Brompton Hospital, London, wrote this cautionary article on heart lung transplantation as it applied to children in the UK and called for open discussion on the subject. His article was published alongside two papers on heart-lung transplantation in children from Great Ormond Street (Whitehead B et al. Arch Dis Child 1991;66;1018-1021 [PubMed] & Whitehead B et al, Arch Dis Child 1991; 66:1022-1026.[PubMed] Of 27 children referred to GOS for assessment of suitability for heart-lung transplant, 10 (37%) were actually transplanted. Six were still alive from three months to three years after the operation but two thirds of the cohort had died at various stages during referral, assessment, and transplant.
Warner maintained that while the transplant has offered miraculous new life to a few children, many more have experienced increased and unnecessary suffering. Planning of transplant programmes must take all facts into account. Also he emphasised that the possibility of heart-lung transplant must not deter further efforts to control chronic lung diseases medically and must not influence appropriate terminal care.
John Warner (fig. 18) was Professor of Child Health at Southampton from 1990 until he moved back to London as Professor of Paediatrics and Head of Department Imperial College St Mary”s hospital campus from September 2006 to April 2013. He continues as Professor of Paediatrics and since 2008 he has been the Director of Research for the Women and Children”s Clinical Programme Group, Imperial College Healthcare NHS Trust (ICHT). He is President of the Academic Paediatrics Association.
These were early days for transplantation (the first adults had only received transplants in 1984), and although there

Fig 18a. Sonny Laing With permission
were some similar views, the latest being in 2008, the outlook for children having transplants continued to improve although as for older patients shortage of donor organs remained a problem.
Sonny Laing (figure 18a). Sonny’s CF was not diagnosed until she was 18 months old when she was referred to Leeds by which time her lungs were severely and irreparably damaged. She was the youngest child with CF to receive a heart-lung transplant at Great Ormond Street, London when she was 5 years old. In the figure she is 15 years old, very well, active and a keen gymnast. Sonny died aged 23 years.
1991 Littlewood AE, Bowler IM, Littlewood JM. A method of data collection and computerisation in use at a regional CF unit. 17th European Cystic Fibrosis Conference Denmark. June 1991. Poster 94.

Fig 19. Ann Littlewood
Much of the clinical research data and the smooth running of the Leeds CF clinic were dependent on the Administrator Mrs Christine Silburn, and the clerks and secretaries.
The computer data management had been in operation since the early Eighties using first a computer funded by the Variety Club of Great Britain.
Ann Littlewood (figure 19) a state registered nurse (and my wife!), was responsible for the running of our data service from the early Eighties until we both retired from clinical work in 1997.
The introduction of a Comprehensive CF Assessment service in Leeds from 1981 generated such vast amounts of data on every patient that computerisation became essential and was introduced in 1982. The system, initially using an Epson PC AX with 80Mb hard disk and 640K using Symantec Q&A software, provided a database and word processing facilities.
By 1991 four databases were in use – a patient register, clinic visit database, an inpatient database and an assessment database for the results of Comprehensive Assessments. The system had been repeatedly modified and adapted over the years by interested colleagues and professional computer programmers.
This early pioneer work eventually progressed and the Leeds CF units are now totally computerised for both clinical and research purposes – a system developed by Professor Daniel Peckham, Respiratory Physician and Director of the Adult CF Unit, St James University Hospital, Leeds.
1991 Mulherin D, Ward K, Coffey M, Keoghan MT, Fitzgerald M. Cystic fibrosis in adolescents and adults. Irish Med J 1991; 84:48-51. 1894494 [PubMed]

Fig. 19a. Muiris Fitzgerald
A cystic fibrosis (CF) clinic for adults in Dublin was established in 1977. The authors reviewed the data on 164 patients who attended between 1977 and 1989. Twenty four patients had died, 11 being over 20 years after time of death. Of the 140 patients still alive, 61% were male and 53% were aged over 20 years. Only 55% were diagnosed by 1 year and 88% by 10 years. Almost all patients had respiratory symptoms and sputum culture yielded Pseudomonas species in 69%. Other respiratory problems included major haemoptysis and pneumothorax, each in 10%. There was a wide range of respiratory impairment among older patients. Among 3 patients aged over 23 years the mean (+/- S.D.) percent predicted FEV1 and FVC were 53.3% (+/- 18%) and 71.4% (+/- 20%) respectively. Mean weight in this group was 92.5% (+/- 14) of predicted. Malabsorption occurred in most patients and meconium ileus equivalent occurred in most patients and meconium ileus equivalent occurred in 34%. Other complications were clinical hepatomegaly (16%), diabetes mellitus (9%) and arthropathy (20%). Most patients were taking continuous antibiotics by mouth (89%) and by nebuliser (48%), beta-2 agonists by inhaler (57%) and oral steroids (29%). Almost all were taking multivitamins, pancreatic replacement therapy and multiple nutritional supplements. The number of CF “bed days” grew 12 fold since 1979 and the mean stay in hospital was double the hospital mean. The economic impact was such that over 1/4 of the annual hospital antibiotic budget was expended on CF patients
This is an account of the situation regarding adults with CF in the Republic of Ireland around this time.
The senior author, Dr Muiris Fitzgerald (figure 19a), was the first adult CF respiratory physician in Ireland at St Vincent’s University Hospital Dublin in 1976. He had trained as a respiratory physician in Ireland, the Unites States and the UK and immediately made a significant impact on CF care in Ireland from then until he retired in 1996. He campaigned actively, as did his successor Professor Charles Gallagher for the adult CF centre at St Vincent’s, although this did not materialise until 2012.
1992 Madden BP, Hodson ME, Tsang V, Radley-Smith R, Khaghani A, Yacoub MY. Intermediate term results of heart-lung transplantation for cystic fibrosis. Lancet 1992; 339:1583-1587. [PubMed]

Fig. 19b Brendan Madden. Spire Healthcare
Between 1984 and 1991 79 patients with CF had heart-lung transplantations in London by Mr Yacoub’s team with a 69% survival to 1 year, 52% to 2 yrs and 49% to 3 years. Cumulative probability of development of obliterative bronchitis was 17%, 23% and 48% at 1, 2, and 3 years. These were encouraging results but the authors noted serious shortage of donor organs – which in 2012 was still a major problem in the UK. (Penketh et al, 1987 above; Yacoub et al, 1990 above; Scott et al, 1988 above for first reports from Brompton and Harefields and Cambridge)
Brendan Madden (fig. 19b) is now a consultant in cardiorespiratory intensive care and respiratory medicine at St Georges Hospital London and at the Royal Brompton Hospital, London.
1992 Handyside AH, Lesko JG, Tarin JJ, Winston RM, Hughes MR. Birth of a normal girl after in vitro fertilization and preimplantation diagnostic testing for cystic fibrosis. N Engl J Med 1992; 327:905-909.[PubMed]

Fig 20. Alan Handyside
Preimplantation genetic diagnosis of cystic fibrosis was attempted in three couples, both members of which carried a delta F508 deletion. In vitro fertilization techniques were used to recover oocytes from each woman and fertilize them with her husband’s sperm. Three days after insemination, embryos in the cleavage stage underwent biopsy and removal of one or two cells for DNA amplification and analysis. Only two oocytes from one woman were fertilized normally; DNA analysis of one of the embryos failed and cystic fibrosis was diagnosed in the other (i.e. it was homozygous for delta F508), so neither was transferred. The oocytes of each of the other two women produced non-carrier, carrier, and affected embryos. Both couples chose to have one non-carrier embryo and one carrier embryo transferred. One woman became pregnant and gave birth to a girl free of the deletion in both chromosomes.
This is the first report of preimplantation genetic diagnosis to identify the delta F508 deletion causing cystic fibrosis using in vitro fertilization, biopsy of a cleavage-stage embryo, and amplification of DNA from single embryonic cells (also Handyside et al, 1988 above; fig.20). Subsequent reports indicated an approximately 30% chance of a successful pregnancy after such an embryo had been implanted.
Professor Alan Handyside (fig. 20) is an internationally-recognised pioneer and expert in human embryology and preimplantation genetics with an extensive research portfolio, track record for innovation in single cell diagnostics.
1992 Anguiano A, Oates RD, Amos JA, Dean M, Gerrard B, Stewart C, Maher TA, White MB, Milunsky A. Congenital bilateral absence of the vas deferens. A primarily genital form of cystic fibrosis. JAMA 1992; 267:1794-1797.[PubMed]
It had been suggested that otherwise healthy men with congenital bilateral absence of the vas deferens (CBAVD), previously considered a distinct genetic entity, have an increased frequency of CF gene mutations (Dumur V et al. Abnormal distribution of CF delta F508 allele in azospermic men with congenital aplasia of epididymis and vas deferens. Lancet 1990; 336:512 [PubMed]; Rigot JJM et al. Cystic fibrosis and congenital absence of the vas deferens. N Eng J Med 1991; 325:64-65.[PubMed] The association was first suggested by Douglas Holsclaw (Holsclaw DS, et al. Congenital abnormalites in male patients with cystic fibrosis. J Urol 1971; 106:568-574.[PubMed]
The present report of 25 unselected men with CBAVD found 16 (64%) had at least one detectable CF mutation, 16 times the expected frequency; 3 men were compound heterozygotes. Some, if not all, otherwise healthy men with CBAVD reflect a newly recognized, primarily genital, phenotype of CF. A very important practical suggestion was that prior to sperm aspiration to remedy infertility, CF mutation analysis should be recommended for them and their partners, as well as for their relatives. In a later study (Chillon M et al. NEJM 1995; 332:1475-1480.[PubMed]) 19 of 102 (18.6%) CBAVD patients had 2 CF mutations and none had the 5T allele. 54 had one copy and 34 had the 5T allele in the other CFTR gene. 29 had no CF mutations but 7 of them had the 5T allele. So most men with CBAVD have mutations in the CFTR gene.
The combination of the 5T allele in one copy of the CFTR gene with a cystic fibrosis mutation in the other copy is the most common cause of CBAVD. The 5T allele mutation has a wide range of clinical presentations, occurring in patients with CBAVD or moderate forms of cystic fibrosis and also in fertile men.
1992 Mennie ME, Gilfillan A, Compton M, Curtis L, Liston WA, Pullen I, Whyte DA, Brock DJH. Prenatal screening for cystic fibrosis. Lancet 1992; 340:214-216. [PubMed]
This is the first report of antenatal couple screening for CF in the Edinburgh maternity hospitals. Of 4348 women, 14% declined prenatal screening and 13% were not screened for other reasons. Amongst 3165 women there were 111 carriers detected of whom four had carrier partners and all 4 couples opted for prenatal diagnosis. One pregnancy with an affected fetus was terminated. The importance of adequate counselling was stressed.
Antenatal screening for CF became routine in Edinburgh but was eventually discontinued in 2005 for various reasons said to include the improving prognosis for CF and also the introduction of neonatal screening in Scotland (also Brock 1985 above; Livingstone et al, 1994 below). National antenatal CF carrier screening had not been introduced in the UK by 2023 although accepted in principle by the UK National Screening Committee. Apparently the cost, particularly the provision of adequate genetic counselling, was a major deterrent. It is unfortunate that such an opportunity is missed but undoubtedly antenatal screening and preconceptional screening will gradually become more common in due course – although it was not routine in the UK by 2023. A definite additional factor is the remarkable change in outlook for people with CF.

Fig. 20a. David Brock
Prof. David Brock was a leading UK researcher who also made major contributions to antenatal diagnosis in of birth neural tube defects and cystic fibrosis.In 1972, Professor David Brock, then a biochemist at the University of Edinburgh, announced that raised amniotic fluid alpha feta-protein (AFP) was associated in pregnancy with open neural tube defects such as spina bifida in the child. Professor Brock was one of the first scientists to apply AFP markers to pre-natal screening. He measured the AFP values in the amniotic fluid of pregnancies with anencephaly or spina bifida. All of the cases of anencephaly and most of those with spina bifida demonstrated markedly elevated AFP levels. This was considered a landmark discovery in the history of pre-natal diagnosis.
1992 Lanng S, Thorsteisson B, Nerup J, Koch C. Influence of the development of diabetes mellitus on the clinical status in patients with cystic fibrosis. Eur J Paediatr 1992; 151:684-687.[PubMed]
The first of a series of important papers about CF related diabetes (CFRD) from Copenhagen and other centres. The new and important message from this paper being that diabetes mellitus adversely affects progress for some time before it becomes clinically obvious. When diabetes develops in CF patients, an insidious decline in overall clinical status is observed for some years prior to its clinical diagnosis. Whether clinical deterioration in CF leads to DM, or pre-diabetes results in declining CF clinical status is unclear. Accumulating evidence suggests that the latter may be the case since insulin therapy seems to improve lung function in cystic fibrosis.
– This Danish study encouraged the introduction of the policy of searching for glucose intolerance when patients of 12 years and older were seen for their Annual Review – a policy agreed by most, but not all, CF physicians.
1992 Conway SP, Simmonds EJ, Littlewood JM. Acute severe deterioration in cystic fibrosis associated with influenza A virus infection. Thorax 1992; 47:112-114.[PubMed]
The role of non-bacterial infection in respiratory exacerbations of cystic fibrosis has been studied less than that of bacterial infection. Some non-bacterial infections, such as influenza A, may be associated with acute respiratory deterioration and may be preventable. Three patients reported here had severe deterioration in their lung function and general wellbeing during the influenza A virus epidemic in the winter of 1989-90.
– Although a Cochrane Systematic Review (Cochrane Database Syst Rev 2009 Oct 7;(4):CD001753.[PubMed]) found no evidence that annual influenza vaccination for people with CF was effective, with experience such as the present report patients with cystic fibrosis are offered immunisation at the beginning of each influenza season. Rapid diagnostic tests and the use of antiviral drugs may have a prophylactic role in minimising lung damage.
1992 Kristidis P, Bozon D, Corey M, Markiewicz D, Rommens J, Tsui LC. Genetic determination of exocrine pancreatic function in cystic fibrosis. Am J Hum Genet 1992; 50:1178-1184.[PubMed]
A review of the association of so-called “mild” mutations with pancreatic sufficiency. Although the majority of CF mutations – including the most common, delta F508 – are strongly correlated with pancreatic insufficiency (PI), approximately 10% of the mutant alleles may confer pancreatic sufficiency (PS). To extend this observation, genomic DNA of 538 CF patients with well-documented pancreatic function status were analyzed for a series of known mutations in their CFTR genes. A total of 30 different, complete genotypes could be determined in 394 (73%) of the patients. The data showed that each genotype was associated only with PI or only with PS, but not with both. This result is thus consistent with the hypothesis that PI and PS in CF are predisposed by the genotype at the CFTR locus; the PS phenotype occurs in patients who have one or two so-called mild CFTR mutations, such as R117H, R334W, R347P, A455E, and P574H, whereas the PI phenotype occurs in patients with two severe alleles, such as delta F508, delta I507, Q493X, G542X, R553X, W1282X, 621 + 1G—- T, 1717-1G—-A, 556delA, 3659delC, I148T, G480C, V520F, G551D, and R560T.
This large study from Toronto strengthens the view that pancreatic function status in CF is genetically determined by specific mutations at the CF locus. Subsequent studies established this beyond any doubt.
1992 Heeley AF, Bangert SK. The neonatal detection of cystic fibrosis by measurement of immunoreactive trypsin in blood. Ann Clin Biochem 1992; 29:361-376.[PubMed]
Anthony Heeley has been a pioneer of neonatal CF screening in the UK. This is a detailed review of the situation at this time (also Green et al, 1993 below for Heeley’s East Anglian results; also Crossley et al, 1979 above).(Also see comments after Crossley & Elliott 1979 above; Heeley et al, 1982 above).
1993 Sheldon CD, Hodson ME, Carpenter LM, Swerdlow AJ. A cohort study of cystic fibrosis and malignancy. Brit J Cancer 1993; 68:1025-8. [PubMed]

Fig 21. Chris Sheldon. author’s photo
A cohort of 412 patients first attending a cystic fibrosis (CF) clinic between 1961 and 1989 were followed up to 30 June 1989. The number of malignancies observed in the cohort was compared with the number expected based on the age, sex and calendar-year-specific cancer registration rates for England and Wales. Four CF patients were diagnosed as having malignancies before 30 June 1989. The tumors were: adenocarcinoma of the terminal ileum; adenocarcinoma of the pancreas, testicular teratoma, and B-cell lymphoma. This compares with 0.89 malignancies expected on the basis of rates in England and Wales (Standardised Registration Ratio = 452; 95% confidence interval 122-1150, P = 0.03). The single case of adenocarcinoma of the terminal ileum contrasts with less than 0.001 expected (P = 0.003) and that of the pancreas with 0.007 expected (P = 0.01). A further adenocarcinoma of the pancreas was diagnosed 2 years after the end of the study period. The two cases of pancreatic cancer compare with 0.008 expected (P = 0.0001) during the period to mid 1991.
On the basis of the present findings and previous case reports in the literature, adenocarcinoma of the pancreas and adenocarcinoma of the terminal ileum may be associated with cystic fibrosis.
This is an early paper from the world’s largest adult CF centre at the Royal Brompton in London suggesting that there may be an association between CF and malignancy – an association later confirmed and particularly associated with CF patients who had received organ transplants. Many of the reports are of isolated case reports.
Dr Chris Sheldon (figure 21) worked at the Brompton Hospital and subsequently became Consultant Pulmonologist and Director of the Adult CF unit at the Royal Devon and Exeter Hospital.
1993 Green MR, Weaver LT, Heeley AF, Nicholson K, Kuzemko JA, Barton DE, McMahon R, Payne SJ, Austin S, Yates JR, et al. Cystic fibrosis identified by neonatal screening: incidence, genotype, and early natural history. Arch Dis Child 1993; 68:464-467.[PubMed]

Fig 22 Michael Green
This is the main report of the East Anglian neonatal CF screening programme. The incidence of cystic fibrosis over the 10 years in East Anglia (a region of the United Kingdom with a population of 2.1 million) had halved. Neonatal screening allowed early diagnosis, genetic counselling of parents and relatives, and more recently the option of prenatal diagnosis in subsequent pregnancies. One hundred and seven children were born with cystic fibrosis between 1981 and 1990, eight of whom were siblings. The Guthrie blood spots of the 82 infants detected by neonatal immunoreactive trypsin screening between 1981 and 1990 were examined for the presence of the most common cystic fibrosis gene mutation (delta F508). It was present in 135 (82%) of the 164 cystic fibrosis genes analysed with 54 (66%) cases being homozygous and 27 (33%) heterozygous. Sixty nine per cent of infants were symptomatic at the time of diagnosis regardless of genotype. No association was found between the early clinical or biochemical features of the disease and homozygosity or heterozygosity for this mutation.Fig 20b
Screening for CF using the blood immunoreactive trypsin assay alone remains an effective method of identifying infants with the CF within the first weeks, thereby allowing early therapeutic intervention. Genetic counselling and prenatal diagnosis have contributed to a reduction in the number of children born with CF, but may not entirely explain the decreasing incidence of the disease (also Heeley AF et al, 1982 above).
Dr. Michael Green (fig. 22) is a consultant paediatrcian in Leicester, UK. Previously he worked in Leeds at the St James CF Unit and in Cambridge. He published a number of papers on nutrition and also on the follow up results of neonatal CF screening in East Anglia.
1993 Walters S, Britton J, Hodson ME. Demographand social characteristics of adults with cystic fibrosis in the United Kingdom. BMJ 1993; 306:549-552.[PubMed]
A survey of adults with CF in the UK by Dr Sarah Walters, (figure 23) a Birmingham doctor who herself has CF, which gave a picture of the relatively new and expanding population of adults with cystic fibrosis in the UK. 1052 adults were members of the Association of Cystic Fibrosis Adults UK,

Fig 23. Sarah Walters
accounting for the majority of adults. Most were living fulfilling lives. 26% of men and 44% of women were married or cohabiting. 55% were working, fewer than 56% had less than two weeks’ sick leave a year. Half of those not employed gave ill health as the reason. Unfortunately, revealing that they had CF at job interviews reduced the likelihood of being employed for those with mild to moderate disease. People with CF had been less successful than the general population in achieving O level or equivalent qualifications, but more successful in achieving A level or higher qualifications.
So contrary to an image of chronic ill health and disability, a high proportion of adults with CF were living full and productive lives – marked contrast to some of the earlier reports of adults with CF.
A further survey by Sarah Walters in 2000 showed considerable further improvement in many aspects of the condition and also the hospital care of the adults with CF in the UK. After a phenomenally active life, Sarah died in 2018.
1993 Govan JR, Brown PH, Maddison J, Doherty CJ, Nelson JW, Dodd M, et al. Evidence for transmission of Pseudomonas cepacia by social contact in cystic fibrosis Lancet 1993; 342:15-19.[PubMed]
Prof. John Govan of Edinburgh (Figure 24) has for many years been a leading UK and international authority on CF microbiology.

Fig 24. John Govan
In this very important paper John Govan reported the definite transfer of B. cepacia infection between people with CF which finally convinced most CF clinicians. An analysis of isolates from 210 patients attending regional CF clinics in Edinburgh and Manchester between 1986 and 1992 showed that the main cause of increased isolations of P. cepacia from 1989 was the emergence of an epidemic strain that had spread between patients in both clinics. Epidemiological evidence indicated that social contact was important in spread of the epidemic strain within and between clinics. Guidelines to limit the acquisition of B. cepacia should not be restricted to patients in hospital, and that intimate or frequent social contact is associated with a high risk of cross-infection.
Following this publication the UK CF Trust advisory group of clinicians and microbiologists advised that strict segregation of B. cepacia-infected patients was essential as some clinics, even in 1993, were not yet segregating B. cepacia infected patients from other CF patients in the clinic. The widespread introduction of segregation from this time led to a steady reduction in the incidence of new B. cepacia infections and a reduction in the prevalence of chronic B. cepacia infections.
Rosenstein & Hall had reported pneumonia and septicemia due to Pseudomonas cepacia in a patient with CF (Rosenstein BJ, Hall DE. Johns Hopkins Med J 1980; 147:188-189.[PubMed]) before the first report from Toronto describing major problems in treating the infection (Gold R et al. J Antimicrob Chemother 1983; 12 Suppl A: 331-336.[PubMed]) and before the other major publication from Toronto (Isles A, et al. Pseudomonas cepaciainfection in cystic fibrosis: an emerging problem. J Pediatr 1984; 104:206-210. [PubMed]above). This Toronto paper describes how the prevalence of Pseudomonas cepacia infection in a population of approximately 500 patients with CF in the Toronto CF centre, increased from 10% in 1971 to 18% by 1981. The carriage of P. aeruginosa had remained unchanged at 70% to 80% over the same period. Patients infected with P. cepacia had greater impairment of pulmonary function than those with P. aeruginosa alone. A syndrome characterized by high fever, severe progressive respiratory failure, leukocytosis, and elevated erythrocyte sedimentation rate (“cepacia syndrome”) had occurred in eight patients over the previous three years, with a 62% fatality rate. Because P. cepacia strains are uniformly resistant to ticarcillin, piperacillin, and aminoglycosides, and because ceftazidime is ineffective despite in vitro activity, treatment of these infections was very difficult. Prevention of acquisition and effective treatment of P. cepacia in patients with cystic fibrosis had become a major problem in the Toronto clinic and this paper describes the devastating effect of the introduction of B. cepacia into a CF clinic population very clearly.
In 1990 we published from Leeds the first UK paper on B. cepacia in CF, having identified 11 patients with the infection over 6 years since 1984 but we found no evidence of obvious cross infection between our patients when we first identified the infection (Simmonds EJ, et al. Pseudomonas cepacia: a new pathogen in patients with cystic fibrosis referred to a large centre in the United Kingdom Arch Dis Child 1990; 65:874-877).[PubMed]
1993 Phillips RJ, Crock CM, Dillon MJ, Clayton PT, Curran A, Harper JI. Cystic fibrosis presenting as kwashiorkor with florid skin rash. Arch Dis Child 1993; 69:446-448. [PubMed]
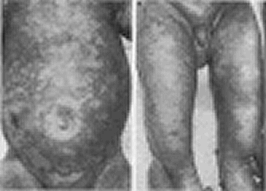
Fig 25. Confluent desquamating rash showing reticulate distribution around islands of normal skin. With permission.
Two infants with a florid erythematous rash (figure 25) and generalised oedema, hypoalbuminaemia, and anaemia were found to have cystic fibrosis. This rare presentation is associated with false negative sweat tests, delays in diagnosis, and a considerable mortality. The authors suggested that this presentation represents a manifestation of kwashiorkor secondary to intestinal malabsorption.
1993 Ranasinha C, Assoufi B, Shak S, Christiansen D, Fuchs H, Empey D, Geddes D, Hodson M. Efficacy and safety of short-term administration of aerosolised recombinant human DNase I in adults with stable stage cystic fibrosis. Lancet 1993; 342:199-202. [PubMed]
A phase II trial of rhDNase (Pulmozyme) from the Brompton Hospital, London, in which the mean percentage change in FEV1 from baseline was a 13.3% rise on rhDNase and only a 0.2% fall on placebo (p < 0.001). There was an insignificant rise of FVC of 7.2% in the rhDNase group and 2.3% in the placebo group.
This study provided further confirmation that short-term administration of rhDNase in stable patients with cystic fibrosis was safe and resulted in an impressive improvement in lung function (also Shak et al, 1990 above; Fuchs et al. 1994 below). The drug proved to be one of the major advances of the decade.
1993 Smith DL, Gumery LB, Smith EG, Stableforth DE, Kaufmann ME, Pitt TL. Epidemic of Pseudomonas cepacia in an adult cystic fibrosis Unit: Evidence of person-to-person transmission. J Clin Microbiol 1993; 31:3017-3022.[PubMed]

Fig 25a. Ty Pitt
Report of transmission between patients in the Birmingham Adult CF Centre investigated in collaboration with Dr Ty Pitt of the Central Public Health Laboratory, London. Prevalence rose from 1.4% in 1988 to 8.3% in 1992. At the time of writing five of the 17 (30%) of the affected patients had died. In only two of the six patients referred to the Birmingham CF centre had P. cepacia been identified before referral.
Dr Ty Pitt (fig. 25a) has been a central figure in understanding the microbiological aspects of cystic fibrosis collaborating closely with clinicians both at the Brompton Hospital in London and also with national studies. Of particular importance was the national survey of CF centres revealing widespread evidence of cross infection both within and between CF Centres in the UK (Scott and Pitt, 2003. below).
1993 Bowler IM, Green JH, Wolfe SP, Littlewood JM. Resting energy expenditure and substrate oxidation rates in cystic fibrosis. Arch Dis Child 1993; 68:754-759.[PubMed]
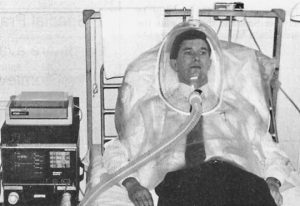
Fig 26. Ian Bowler during the study
Ian Bowler (fig. 26) coordinated this study on the resting energy expenditure (REE) and substrate oxidation rates in 16 patients with CF who had mild chest disease. Eleven healthy controls were measured using indirect calorimetry. The mean REE (% predicted) in the patients with CF was 11% greater than in the controls. Five patients with CF were hypermetabolic but only one of these had a clinically significant reduction of respiratory function. A greater proportion of REE was derived from carbohydrate oxidation in the CF patients (43.5% v 29.9%). However, the 24 hour dietary intake of carbohydrate was greater in the cystic fibrosis group (49.6 v 45.8% of energy intake). These data suggest that a high dietary intake of carbohydrate may contribute to the increased oxidation of carbohydrate in these CF patien
Ian Bowler at this time was our CF Research Fellow in Leeds. He subsequently became consultant paedaitrian in Newport, Wales where he organised the CF services.
1993 Littlewood JM. The value of comprehensive assessment and investigation in the management of cystic fibrosis. In Clinical Ecology of Cystic Fibrosis. H Escobar, CF Baquero Suarez (Eds). Elsevier Science Publishers. 1993:181-187.[Conference publication]
This is a report of the 427 new patients seen at the Leeds CF centre for a Comprehensive CF Assessment between 1980 and 1992, 364 of whom had at least one follow-up assessment. They were referred from 36 hospitals (10 in Yorkshire and a further 26 hospitals in the UK). The details of their management and condition are recorded. A comparison is made between a previous 100 patients referred between 1985 and 1987 and 100 new referrals between 1988 and 1992 showing improvement in their condition in a number of areas reflecting the general improvement in CF care in the UK (figure 27). The details give an idea of the condition of the patients at the time. See also Littlewood et al, 1984 above; Littlewood et al, 1988 above)
1994 Fuchs HJ, Borowitz DS, Christiansen DH, Morris EM, Nash ML, Ramsey BW, Rosenstein BJ, Smith AL, Wohl ME. Effect of aerosolized recombinant human DNase on exacerbations of respiratory symptoms and on pulmonary function in patients with cystic fibrosis. The Pulmozyme Study Group. N Engl J Med 1994; 331:637-642. [PubMed]

Fig 28. Henry Fuchs
Dr Henry J Fuchs (fig. 28), while working at Genentech from 1987 to 1996, led the clinical programme that resulted in the approval of Pulmozyme – one of the major clinical advances introduced during the Nineties. Dr Fuchs is now Senior Vice President and Chi ef Medical Officer of Biomarin.
This report, the main publication on rhDNase (Pulmozyme) describes a randomized, double-blind, placebo- controlled study to determine the effects of once-daily and twice- daily administration of rhDNase on frequency of exacerbations of respiratory symptoms requiring parenteral antibiotics and on pulmonary function.
A total of 968 adults and children with cystic fibrosis were treated for 24 weeks as outpatients. As compared with placebo, the administration of rhDNase once daily and twice daily reduced the age-adjusted risk of respiratory exacerbations by 28 percent (P = 0.04) and 37 percent (P < 0.01), respectively. The administration of rhDNase once daily and twice daily improved forced expiratory volume in one second during the study by a mean (+/- SD) of 5.8 +/- 0.7 and 5.6 +/- 0.7 percent, respectively. Transient voice alteration and laryngitis were more frequent in the rhDNase-treated patients.
This was the definitive Phase III trial showing the significant effect of inhaled rhDNase – one of the most important large multicentre studies of the 1990s. Pulmozyme was undoubtedly one of the major clinical advances of the decade and was licensed by the FDA in 1994 following this study. Regular treatment with DNase would become widely used (by 2021 86% of people over 6 years old, 43.6% less than 3 years and 69% 0f 3-5 year olds with CF on the CF Foundation registry were taking regular DNase) first amongst those with significant chest involvement and then later for those with milder chest problems (Quann et al, 2001 below) and eventually, in some CF units for very young infants. The cost (approximately £7000 per year in the UK) at the time proved a problem in certain parts of the UK. (Also Shak et al, 1990 above; Ranasinha et al, 1993 above).
1994 Sawyer S, Bowes G, Phelan PD. Vulvovaginal candidiasis in young women with cystic fibrosis. BMJ 1994; 308:1609. [PubMed]
Vulvovaginal candidiasis was more common in 55 women with CF than in controls (13 vs.4) and more difficult to treat. Many women with CF had recognized the association of the Candida infection with their use of antibiotics. The authors suggested women with CF should be given routine advice about the possibility of candidiasis.
This was an important paper as it is unlikely that women would be asked about such problems in a busy CF clinic for adults which are often “chest orientated” – yet adequate treatment of the candidiasis would significantly improve the patient’s quality of life. Somewhat analogous to this problem was the later recognition of the increased incidence of urinary incontinence in women with CF (see Cornacchia et al, 2001).
1994 Super M, Schwarz MJ, Malone G, Roberts T, Haworth A, Dermody G. Active cascade screening for carriers of cystic fibrosis gene. BMJ 1994; 308:1462-1467. [PubMed]

Fig 29. Maurice Super
Dr Maurice Super (1936-2006) (fig. 29) first encountered CF in Windhoek in South West Africa (Namibia) in 1967 where he started a CF clinic. He subsequently became a leading geneticist and paediatrician in the UK working in Manchester. He was a major protagonist of carrier screening in the extended families of people with cystic fibrosis – so-called “cascade screening”.
The present paper describes 15 carrier couples detected out of 1563 relatives of people with CF who were tested; eight had prenatal tests and three pregnancies were terminated. An average of 16 people per family had been tested. Cascade screening was acceptable to relatives, particularly on the mother’s side of the family and 10 times more successful in detecting carrier couples than unfocused screening. The genetic testing of all child-bearing relatives of a person with CF is now provided by the UK NHS if the individuals wish to be tested.
1994 Arens R, Gozal D, Omlin KJ, Vega J, Boyd KP, Keens TG, Woo MS. Comparison of high frequency chest compression and conventional physiotherapy in hospitalized patients with cystic fibrosis. Am J Res Crit Care 1994; 150:1154-1157.[PubMed]
A study of 50 people with CF admitted for acute pulmonary exacerbations that were randomly allocated to receive either high frequency chest compression (HFCC) or conventional physiotherapy (CPT) three times a day. After seven and 14 days of treatment, improvements were similar in the two study groups, leading to patient discharge after similar periods of hospitalization.
The authors concluded that HFCC and CPT are equally safe and effective when used during acute pulmonary exacerbations in CF patients. They suggested that HFCC may provide an adequate alternative in management of CF patients in a hospital setting. (Also Warwick WJ, Hansen LG, 1991 above)
1994 Livingstone J, Axton RA, Gilfillan A, Mennie M, Compton M, Liston WA, Liston WA, Calder AA, Gordon AJ, Brock DJ. Antenatal screening for cystic fibrosis: a trial of the couple model. BMJ 1994; 308:1459-1462. [PubMed]
The second report of antenatal screening of 8536 couples in Edinburgh. 8.4% were “ineligible”, 1900 declined screening for various reasons and 5922 (69.4%) were screened. There were four positives (i.e. both partners were CF carrier heterozygotes) and all four couples opted for prenatal diagnosis. There were three terminations where the fetus was affected and one couple elected to have the CF infant. There was 99% satisfaction by those screened.
Antenatal CF screening was pioneered in Edinburgh by David Brock and his colleagues and this is one of the first reports (also Mennie et al, 1992 first report above). Screening was introduced into the two Edinburgh trial hospitals following this report. However, the service was eventually discontinued in 2005 soon after neonatal CF screening was introduced into Scotland. As the outlook for CF improved parental and general attitudes changed to antenatal diagnosis and termination, also the mutations tested differed from the neonatal ones, and finally funding for both antenatal and neonatal screening was inadequate. It has been estimated from various studies that for every CF fetus detected by antenatal screening the cost is between £50K and £100K.
1994 Walters S, Britton J, Hodson ME. Hospital care for adults with cystic fibrosis: an overview and comparison between special cystic fibrosis clinics and general clinics using a patient questionnaire. Thorax 1994; 49:300-306.[PubMed]
Another study from Sarah Walters on this occasion to assess the current pattern of medical service received by adults with CF in the UK and to compare the type of care between special CF centres and general clinics. Two thirds of the patients were attending special CF centres for either adults or adults and children. Significant differences between cystic fibrosis and general clinics were noted. Patients attending special cystic fibrosis clinics were more likely to have had simple clinical investigations (blood tests, sputum culture, oxygen saturation, chest radiography, weight and lung function measurement) in the previous year. They were also more likely to have received intravenous antibiotics at home, and to have access to paramedical personnel. Patients attending cystic fibrosis clinics were taking higher doses of pancreatic enzyme supplements with respect to quantity and potency of preparation. Such patients also had less severe symptoms irrespective of social class, and were more likely to be satisfied with professional aspects of their care. Regardless of type of clinic, potential deficiencies were identified in overall medical care with omission of clinical investigations in severely affected patients and evidence of under treated respiratory and digestive symptoms in patients with moderate and severe disease.
This survey provides evidence that adults with cystic fibrosis attending special cystic fibrosis clinics at that time received more intensive care, had better symptom control, and are more satisfied with the service provided than those attending general chest clinics. The introduction of CF centre care was slow in the UK and was opposed by some consultants working in general hospitals with a few CF patients. Even in 2008 there was still considerable discussion about the best care arrangements for people with CF. It is recommended by all CF organisations, including the UK CF Trust, that all adults with CF should receive all their medical care at a specialist CF Centre for adults.
1994 Ozçelik U, Göçmen A, Kiper N, Coşkun T, Yilmaz E, Ozgüç M .Sodium chloride deficiency in cystic fibrosis patients. Eur J Pediatr. 1994;153(11):829-31. [PubMed]

Fig 30. Ayhan Gocmen Author’s photo
Sodium chloride deficiency (SCD) was observed within the 1st year of life in 12 of 46 cystic fibrosis (CF) patients between July 1989 and September 1992. All patients showed sweating, loss of appetite, fever, vomiting, irritation, dehydration, weakness, and cyanosis during an attack. Mean plasma sodium, potassium and chloride levels were 122.9 (range 106-135), 2.5 (range 1.6-3.5), and 73.3 (range 60-90) mEq/l respectively. Alkalosis and elevated plasma renin activity were detected in all patients. Interesting full details in the abstract.
Another example of “pseudo-Bartter’s syndrome” which is undoubtedly a more common occurence in hot climates such as Turkey.
Professor Ayhan Göçmen (fig. 30) of Ankara has been involved in CF care and research in Turkey for many years since the mid-Seventies. In 2007 he was President of the ECFS Conference in Belek, Turkey.
1994 Weaver LT, Green MG, Nicholson K, Mills J, Heeley A, Kuzemko JA, et al. Prognosis in cystic fibrosis treated with continuous flucloxacillin for the neonatal period. Arch Dis Child 1994; 70:84-89.[PubMed]

Fig. 31 Lawrence Weaver. universtystory.gla.ac.uk
This study from East Anglia provided satisfactory evidence for most people in the UK that regular prophylactic flucloxacillin during the first two years in 38 screened CF infants was associated with less cough, fewer Staphylococcus aureus isolates, a lower hospital admission rates (19 versus 5) and for shorter periods (6.4 versus 2.2 days) and the need for only half the number of additional antibiotics. The study was possible as neonatal screening had been introduced into East Anglia by Anthony Heeley and his colleagues in 1981 – the first IRT screening programme in the UK ( Heeley et al, 1982 above).
Prophylactic anti-Staphylococcal therapy had been recommended many years ago by David Lawson, paediatrician at Carshalton, London and was routine practice in some CF centres in the UK, from the mid-Seventies.
Evidence from this trial provided a basis for the CF Trust Antibiotic Group’s recommendations in 2002 (and confirmed later in the 2009 edition) for all infants with CF to receive continuous flucloxacillin for the first 2 years; in fact the first 3 years was recommended in 2009.
Discussion continued for the next decade as to whether this treatment increased the likelihood of Pseudomonas infection – this was not really relevant in UK centres where early effective eradication of Pseudomonas had been practised since the early Eighties but probably was the case in the USA where early eradication of Pseudomonas was not yet routine practice.
It should be noted that as an alternative to long term flucloxacillin, the incidence of chronic S. aureus infection could also be reduced by early active antibiotic treatment whenever the organism was cultured as shown in Copenhagen (Szaff & Hoiby, 1981 above). It is important to note that in Copenhagen monthly respiratory cultures are performed so S. aureus is always detected at an early stage when it can be eradicated; as with P. aeruginosa, once allowed to become chronic eradication is difficult if not impossible.
The very high prevalence of chronic S. aureus infection reported in recent CFF Registry figures (in up to 60% by 2yrs and 80% of patients by the age of 10yrs) and from a number of countries, including the UK, is surprising considering that S. aureus was the main cause of death in the early years and is still associated with a definite inflammatory response as shown in bronchoscopic studies of young CF infants (Armstrong et al, 1995 below).
Professor Lawrence Weaver (figure 31) is Professor of Child Health in Glasgow (1996-2011) and paediatrician at the Royal Hospital for Sick Children Glasgow. This research included the neonatal screened infants from East Anglia, which was carried out when he was working at the MRC Dunn Nutrition Unit Subsequently Lawrence Weaver’s interests have turned to the history of biology and medicine, art and architecture. You can find more about him at lawrencetrevelyanweaver.com
1994 Green MR, Weaver LT. Early and late outcome of cystic fibrosis screening. J R Soc Med 1994; 87 (Suppl 21): 5-10.[PubMed]
More data from the East Anglian screened CF infants. During the decade although the birth rate in the region had increased, the incidence of CF had halved. Early detection of CF by neonatal screening allows genotyping of infant and family. It also offers parents of an affected child the opportunity of counselling before embarking on further pregnancies and if desired subsequent antenatal testing. So both neonatal and antenatal CF screening (Cunningham et al, 1998 below) appear to reduce thee incidence of CF. This has been the experience in a number of regions but not all – for example Colorado.
Dr. Michael Green (fig. 22 above) is a consultant paediatrcian in Leicester, UK. Previously he also worked in Leeds at the St James CF Unit and in Cambridge. He published a number of papers on nutrition and also on the follow up results of neonatal CF screening in East Anglia.
1994 Smyth RL, van Velzen D, Smyth AR, Lloyd DA, Heaf DP. Strictures of the ascending colon in cystic fibrosis and high strength pancreatic enzymes. Lancet 1994; 343:85-86. [PubMed]
The first report of fibrosing colonopathy – a new, unexpected and serious complication subsequently shown to be related to the use of the new high
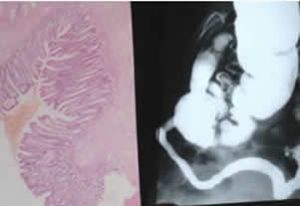
Fig 33. Fibrosing colonopathy. barium enema showing severe stricture (R) and histology of the colon with extensive fibrosis.
strength pancreatic enzyme preparations that had been introduced during the previous 2 years (Pancrease HL, Creon 25,000, Nutrizym 22). The authors observed five children with CF, who presented over a period of two months, with meconium ileus equivalent that failed to respond to medical management. At surgery, four had a stricture in the ascending colon (figure 32 is similar but less severe), and all had histopathological changes of post-ischemic ulceration repair, with mucosal and submucosal fibrosis (figure 33). The only common change in the management of these children had been a switch from conventional enteric-coated pancreatic enzymes to a high-strength product between 12 and 15 months before presentation.
A subsequent survey of 114 CF care centres in the United States revealed 15 cases of colonic stricture all of whom had been taking relatively large doses of the new enzymes (6,700 to 29,100 units /lipase/kg per meal)(Freiman JP, FitzSimmons SC. Colonic strictures in patients with cystic fibrosis: results of a survey of 114 cystic fibrosis care centers in the United States. J Pediatr Gastroenterol Nutr 1996; 22:153-156. [PubMed])
A case controlled study in the UK from Smyth et al, 1995 (below) showed a significant correlation with both a high enzyme dose and the make of preparation – only those enzymes containing a covering of the copolymer eudragit being implicated (Pancrease HL and Nutryzim 22 but not Creon 25,000). A similar study from the USA confirmed the association with high enzyme dosage but not with the copolymer covering or brand (Fitzsimmons SC et al. High dose pancreatic enzyme supplements and fibrosong colonopathy in children with cystic fibrosis. N Eng J Med 1997; 336:1289-9).
However, no further cases were reported in the UK or have been since reported, in patients taking the high strength Creon 25,000 preparation (which does not contain eudragit) even when given to children for 3 years in high doses (Connett et al, 1999 below).
Fig 32. partial fibrosing colonopathy showing narrowing of the ascending colonwhich did not progress after the high strength enzyme (Pancrease HL) was replaced by Creon
In the UK the recommendations of the Committee on Safety of Medicines to avoid doses of enzymes in excess of 10,000 IU lipase/kg/day and preparations containing the copolymer eudragit in children, abolished the condition in the UK although a few cases continued to occur in the USA. The enzyme content of the various preparations are described in detail elsewhere (Littlewood JM, Wolfe SP, Conway SP. Diagnosis and treatment of malabsorption in cystic fibrosis. Pediatr Pulmonol 2006; 41:35-49.[PubMed]
1994 Wilfond BS, Farrell PM, Laxova A, Mischler E. Severe hemolytic anemia associated with vitamin E deficiency in infants with cystic fibrosis. Implications for neonatal screening. Clin Pediatr 1994; 33:2-7. [PubMed]
Three infants with CF and malnutrition leading to severe anemia beginning as early as six weeks of age. They had high reticulocyte counts, negative Coombs’ tests, abnormal peroxide haemolysis test results, and biochemical evidence of vitamin E deficiency. Oral administration of alpha-tocopherol resulted in rapid correction of the laboratory abnormalities.
Phillip Farrell of Wisconsin has published extensively on vitamin E deficiency both in premature infants and those with CF (see Farrell et al, J Clin Invest 1977; 60:233-241 above). This report emphasises the early onset of fat soluble vitamin deficiencies in infants with CF as also found in the Colorado screened infants (Sokol et al, 1991 above).
1994 Conway SP, Pond MN, Bowler I, Smith DL, Simmonds EJ, Joannes DN, Hambleton G, Hiller EJ, Stableforth DE, Weller P, Littlewood JM. The chest radiograph in cystic fibrosis: a new scoring system compared with the Chrispin-Norman and Brasfield scores. Thorax 1994; 49:860-862. [PubMed]
A chest X-ray score devised and evaluated by members of the Northern CF Club (NCFC) (figure 34) – an informal group of senior paediatricians and respiratory physicians who were treating increasing numbers of children with CF in the North of England. The group was formed and chaired in the mid-Eighties by Jim Littlewood to discuss difficult clinical problems encountered with increasing frequency by those of us responsible for treating people with cystic fibrosis. The initial format of the meeting was to meet at the Cottons Hotel in Cheshire to discuss specific patients during the late afternoon and evening. It was essentially a mutual support group for clinicians to help each other to decide the best treatment for some of these problems which many of us were encountering for the first time.

Fig 34. Some of the original members of the Northern CF Club outside The Cottons Hotel in Knutsford Cheshire.
“Northern X-ray Score” is now used routinely in many CF centres in the UK and the authors suggested that it – “fulfils the requirements of a chest radiograph score more successfully than the Chrispin-Norman or Brasfield systems and does not require a lateral film” – hence reducing considerably the irradiation to the patient over the years. So the published research output of our Northern CF Club was quality not quantity! However, countless patients have benefitted from the advice their doctors received from their colleagues at these NCFC meetings.
1994 Beardsmore CS, Thompson JR, Williams A, McCardle EK, Gregory GA, Weaver LT, Simpson H. Pulmonary function in infants with cystic fibrosis: effect of antibiotic treatment. Arch Dis Child 1994; 71:133-137.[PubMed]
Children from the East Anglian neonatal screening programme (1985-1992) who were included in the flucloxacillin trial (Weaver et l, 1994 above) underwent infant respiratory function tests at three to four months and one year of age (measurements of thoracic gas volume and airway conductance using an infant whole body plethysmograph and maximum expiratory flow by the “squeeze” technique). There was no difference in lung function between the flucloxacillin treated infants and the control CF infants at any age.

Fig 35. Caroline Beardsmore
Dr Caroline Beardsmore (figure 35) was one of the few experts in infant lung testing at the time in the UK, eventually Senior Lecturer in the Department of Respiratory Sciences and the Biomedical Research Centre (Respiratory theme), University of Leicester, Leicester, UK. Department of Paediatric Respiratory Medicine, Leicester Children’s Hospital, University Hospitals Leicester, Leicester, UK.
Periodically studies are published on infant respiratory function testing but the techniques are so complicated and time-consuming that they are usually only performed by the authors of the papers. Although valuable for research they have never been of great practical value in the clinic for most patients. However, as early intervention is now regarded as of increasing importance the measurement of neonatal respiratory function becomes essential to monitor the results of the interventions.
In the present study it is not surprising that there was no difference between the treated and control infants as some hyperinflation is present in all CF infants from early infancy whether they are infected or not. Some degree of hyperinflation in most CF infants was confirmed in subsequent studies (Ranganathan et al, 2001 below).
1994 Konstan MW, Hilliard KA, Norvell TM, Berger M. Bronchoalveolar lavage findings in cystic fibrosis patients with stable clinically mild lung disease suggest ongoing infection and inflammation. Am J Respir Crit Care med 1994; 150:448-454. [PubMed]
A rather surprising study on 18 patients which appears to demonstrate the obvious i.e. that bronchiolar alveolar lavage in adolescents, who were all chronically infected with P. aeruginosa, S. aureus and/or H. influenzae reveals evidence of ongoing inflammation. It would have been surprising if it had not revealed evidence of inflammation! The authors conclude “there is significant ongoing infection and inflammation in the airways of CF patients with clinically mild lung disease (FEV1 79%+/- 4%) and suggest a more aggressive intervention might preserve their lung function for few of these patient had received recent IV antibiotics (nine never and five not in the past three years) and there is no mention of their taking nebulised antibiotics.
The unfortunate use of the term “colonised” is evident from this study – repeated positive cultures indicates that there is certainly chronic “infection” of tissue involvement rather than the mere presence of organisms within the airways. This would have been evident from serum antibody studies and by the presence of inflammatory markers in the airways. The authors’ conclusions that “intervention aimed at reducing ongoing infection and destructive inflammatory response might be beneficial even when patients do not have signs and symptoms of acute exacerbations” – seems obvious and is a policy already adopted by the Danish CF centre since the early Eighties and by many CF centres in the UK and Europe for many years.
1995 Gan K-H, Geus WP, Bakker W, Lamers CBHW, Heijerman HGM. Genetic and clinical features of patients with cystic fibrosis diagnosis after the age of 16 years. Thorax 1995; 50:1301-1304.[PubMed]
Predictably, adult patients with late diagnosis have a better prognosis. The differences between the early diagnosis of 4.6 years (ED) and late diagnosis 27.7 years (LD) individuals were FEV1 52% vs 72.5%; Pseudomonas infection in 70% vs 24%; pancreatic insufficiency 81% vs 12%; homozygous for DF508 58% vs none.
In other series of adults with CF many have been diagnosed late and had other features suggesting a significant number had milder mutations – supported by the high frequency of pancreatic sufficiency. However, it has been emphasised that these patients require just as careful follow up and treatment as those with the more usual clinical picture – rather than waiting until they develop more obvious chest symptoms (Lording A, et al. Pulmonary infection in mild variant cystic fibrosis: implications. J Cyst Fibros 2006; 5:101-104. [PubMed]
1995 Gan KH, Veeze HJ, van den Ouweland AM, Halley DJ, Scheffer H, van der Hout A, Overbeek SE, de Jongste JC, Bakker W, Heijerman HG. A cystic fibrosis mutation associated with mild lung disease. New Eng J Med 1995; 333:95-99. [PubMed]
Among Dutch patients A455E is the second most common mutation and associated with preserved pancreatic function and some residual secretion of chloride across membranes. Thirty three patients with compound heterozygosity for the A455E mutation were compared with matched controls homozygous for the delta F508 mutation. In those with the A455E mutation diagnosis was later (mean age at diagnosis, 15.0 vs. 3.1 years; P < 0.001);

Fig 36. Harry Heijerman
fewer had pancreatic insufficiency (21.2 percent vs. 93.9 percent, P < 0.001), and none had diabetes mellitus (0 percent vs. 27.3 percent, P = 0.004). FEV1 and FVC were significantly higher (FEV1, 73.9 vs. 54.3 % predicted P = 0.002) and FVC, 88.7 vs. 76.3 % predicted P = 0.04). Fewer had chronic Pseudomonas infection (33.3 vs. 60.6 % P = 0.02).
Although several mutations were known to be associated with less severe pancreatic disease, these findings showed a correlation between the A455E mutation and mild pulmonary disease resulting in a better prognosis.
Dr Harry Heijerman (figure 36) was the Founding Editor of the Journal of Cystic Fibrosis. Under his guidance the journal was increasingly successful; the editorship was taken over by Prof. Gerd Döring in 2006 (see also 2002 Journal of Cystic Fibrosis below).
Harry Heijerman is Physician at Haga Teaching Hospital, Deb Haag in the Netherlands and a leading figure in CF care and research in Europe.
1995 The Cystic Fibrosis and Genetic Disorders Group is a Collaborative Review Group (CRG) of The Cochrane Collaboration

Fig 37. Ros Smyth
This Cochrane group for CF was formed in 1995 and was expanded to cover other genetic disorders in 1997. It consists of a team led by Prof. Ros Smyth of Liverpool (figure 37) who are interested in producing high quality Systematic Reviews of controlled clinical trials in CF and, since 1997, other genetic disorders.
Initially supported by the UK CF Trust, the group is now supported by the NHS R&D (UK). Reviewers undertake the work on a voluntary basis and by 2014 there were 152 Systematic Reviews on various aspects of treatment and management. Almost all the reviews highlight the lack of controlled trials on many aspects of treatment; however, the information from systematic reviews published on the website has proved an increasingly valuable source of information for those dealing with CF – even though the stringent standards of the Reviewers on occasion have proved irritating to some elderly clinicians!

Fig 38. Archie Cochrane
Undoubtedly Ros Smyth and the Group have had a major and favourable influence on the number and standards of clinical trials which have influenced the management of people with CF.
Professor Archie Cochrane (1908-1988) (figure 38) was born in Kirklands, Galashiels, Scotland. He qualified in 1938 at University College Hospital, London, and joined the Medical Research Council’s Pneumoconiosis Unit at Llandough Hospital, a part of Cardiff University School of Medicine in 1948. Here he began a series of studies on the health of the population of Rhondda Fach — studies which pioneered the use of randomised controlled trials. His 1971 Rock Carling monograph ‘Effectiveness and Efficiency: Random Reflections of Health Services’ was very influential. These ideas and his advocacy of randomized controlled trials eventually led to the development of the Cochrane Library database of systematic reviews, the establishment of the UK Cochrane Centre in Oxford and the international Cochrane Collaboration (from Cochrane website).
1995 Armstrong DS, Grimwood K, Carzino R, Carlin JB, Olinsky, Phelan PD. Lower respiratory infection and inflammation in infants with newly diagnosed cystic fibrosis. BMJ 1995; 310:1571-1572.[PubMed]
An important study from Melbourne documenting the early onset of infection in screened infants with CF who, incidentally, were not treated with long term anti-Staphylococcal antibiotics. Forty five infants (32 screened and 12 with meconium ileus) had bronchoalveolar lavage at a mean age of 2.6 months. Sixteen (36%) already had respiratory symptoms and seven were receiving antibiotics at the time, although long term flucloxacillin was not routine policy. Already lower respiratory infection, usually with S. aureus, was present in almost 40% (17/45) of these young CF infants of whom a third were symptom free. Follow up showed P. aeruginosa to be present in some infants as early as 4 months.
— I believe this study lends strong support to the use of long term anti-Staphylococcal flucloxacillin, at least for the first 2 or 3 years as recommend by the UK CF Trust’s Antibiotic Group in both 2002 and 2009 (full text on CF Trust website); also this policy is supported by the trial of Weaver et al.1994 (above).
1995 Bowler IM, Kelman B, Worthington D, Littlewood JM, Watson A, Conway SP, Smye SW, James SL, Sheldon TA. Nebulised amiloride in respiratory exacerbations of cystic fibrosis: a randomised controlled trial. Arch Dis Child 1995; 73:427-430. [PubMed]

Fig. 38a Ian Bowler
As a result of Michael Knowles’s amiloride trial (Knowles et al, 1990 above), in Leeds we assessed the benefit of nebulised amiloride added to the standard treatment of a respiratory exacerbation in people with cystic fibrosis.
Dr Ian Bowler (Fig. 38a), our Leeds CF Research Fellow at the time, performed a prospective, randomised, double blind, placebo controlled trial with 27 patients (mean age 12.8 years) in two hospitals in Leeds, UK. Both forced expiratory volume in one second (FEV1) and forced vital capacity (FVC) showed improvements over the course of treatment as would be expected but there was no difference in respiratory function between the two groups at any of three time periods during the study. However, the time to reach peak FVC was significantly reduced in the amiloride group (4.2 v 7.6 days; 95% CI 0.4 to 6.4 days), but not in the time to reach peak FEV1 (5.7 v 7.9 days; 95% CI -1.2 to 5.6 days). Amiloride did not result in a greater overall improvement in respiratory function.
On the modest results of this trial we did not introduce amiloride into the treatment regimen for exacerbations. No benefit was seen from amiloride by Graham A et al (No added benefit from nebulised amiloride in patients with cystic fibrosis. Eur Respir J 1993; 6:1243-1248. [PubMed]) nor in a later French multi-centre randomized double blind placebo controlled trial in patients more than 5 years old (Pons G, et al, Pediatr Pulmonol 2000; 30:25-31. [PubMed]).
However, earlier Kohler et al showed inhaled amiloride improved mucociliary clearance in patients with CF (Kohler et al, Eur J Respir Dis 1986; 69 Suppl 146:319-326); also the same group confirmed this with a larger study (App AM et al. Am Rev Respir Dis 1990; 141:605-612. [PubMed]). Also Lindemann et al from Giessen had reported 50.4% more sputum was produced by autogenic drainage after inhalation of amiloride than after isotonic saline also visible liquefaction of secretions was noted by the physiotherapist and patients (Lindemann H et al. Elimination of secretions in CF patients under amiloride inhalation. Pneumologie 1990; 44:1148-1150 [PubMed] [German]).
Later more active and longer acting drugs (P- 680 & P- 522-O2-Parion Sciences then Gilead who renamed P-680 as GS9411) that inhibit excess Na absorption showed more promise but eventually proved ineffective in Phase III trials. Apparently, the duration of action of amiloride is so very short to the extent that it would be unlikely to have a significant beneficial effect.
1995 Smyth RL, Ashby D, O’Hea U, Burrows E. Lewis P. van Velzen D. Dodge JA. Fibrosing colonopathy in cystic fibrosis. Results of a case controlled study. Lancet 1995; 346:1247-1251. [PubMed]
Fibrosing colonopathy was first described in children with CF in 1994 by Dr (now Prof.) Ros Smyth (figure 34 above) and her colleagues. This study confirmed the relationship between fibrosing colonopathy and high doses of lipase and the relation of fibrosing colonopathy with certain brands of high strength enzymes, which contained a copolymer – Eudragit. This is a report of a nested case-control study to identify possible associations with this condition. A case ascertainment within the UK CF population to identify any cases that had occurred between January, 1984, and April, 1994, revealed 14 cases, all aged less than 14 years and all confirmed by independent histopathological review. All had presented since April, 1993; 12 were boys and six had received some or all of their care in Liverpool. Each child was matched, by date of birth, with four controls from the UK CF Registry. Information was obtained about the cases and the controls from their case records and by a structured interview with the families.
In the 12 months before surgery, there was a dose related association between the occurrence of fibrosing colonopathy and use of high-strength pancreatic enzyme preparations. Odds ratio per extra 1000 high-strength capsules was 1.45 (95% CI 1.14-1.84). For use of protease, the odds ratio per million extra units per kg was 1.55 (1.19-2.03). For usage of individual high-strength products at any time during the 12 months before surgery some differences were observed; for Creon 25000 the odds ratio was 0.38 (0.10-1.42), for Nutrizym 22 it was 43.4 (2.51-751), and for Pancrease HL 8.4 (1.95-36.1). These last two confidence intervals are extremely wide and compatible with these two products having the same odds ratios. Laxative use was independently predictive (odds ratio 2.42 [1.20-4.94]).
The authors concluded that there was a definite dose-related association between high-strength pancreatic enzyme preparations and fibrosing colonopathy. (see also Fibrosing colonopathy in children with cystic fibrosis. Proceedings of a conference in Manchester organised by the Cystic Fibrosis Trust, 5th Nov. 1995. Littlewood JM, Hind CRK (eds). Postgrad Med J 1996; 72 (Suppl 2):S1-S64).

Fig 39. Deborah Ashby
Professor Deborah Ashby OBE (fig. 39) holds the Chair in Medical Statistics and Clinical Trials at Imperial College London. She has been involved with number of cystic fibrosis projects including the original Clinical Trials Group of the Cystic Fibrosis Trust, also in the present studies relating to fibrosing colonopathy, in various other clinical trials and with the Reviews for the Cochrane Cystic Fibrosis and Genetic Disorders Group with Professor Ros Smyth. Deborah was awarded the OBE for services to medicine in 2009.
1995 Pamucku A, Bush A, Buchdahl R. Effects of Pseudomonas aeruginosa colonisation on lung function and anthropomorphic variables in children with cystic fibrosis. Pediatr Pulmonol 1995; 19:10-15.[PubMed]
Sixty two children with CF had three or more Annual Assessments of their lung function at the Brompton Hospital, London. Despite optimal pulmonary management children who were chronically infected with P. aeruginosa deteriorated significantly faster than those not so infected.
The term “chronically infected” would have been preferable to the term “colonised” used in this paper. This is one of a number of valuable studies confirming an increasing rate of pulmonary deterioration following the onset of chronic Pseudomonas infection – an event which has been aptly described as “The point of no return” (Drittanti et al,1997 below). Also considered a serious matter by the late Cristian Koch in 1997 when he said – “When I look back on what we’ve done all through the years that I’ve been involved with cystic fibrosis, I would say that the early treatment of Pseudomonas is probably the best thing that we have done for the patients. It becomes more and more clear that really what determines the long term course is whether you get Pseudomonas or not” .
There are other studies confirming the importance of avoiding chronic Pseudomonas infection (Kerem E et al, J Pediatr 1990; 116::714-719. [PubMed]; Henry RL et al. Pediatr Pulmonol 1992;12:158-161[PubMed]; Hudson VL et al. J Pediatr 1993; 122:854-860.[PubMed]; CF Foundation Registry, 1996; Frederiksen B et al. Pediatr Pulmonol 1997;23:330-335.[PubMed]; Kosorok MR et al, Pediatr Pulmonol 2001; 32:277-287.[PubMed]).
1995 Konstan MW, Byard PJ, Hoppel CL, Davis PB. Effect of high dose ibuprofen in patients with cystic fibrosis. N Engl J Med 1995; 332:848-854.[PubMed]

Fig 40. Mike Konstan
This was a major study funded by the CF Foundation to determine the benefits of treating inflammation in the CF airways with long term oral ibuprofen. A double-blind trial involving 85 patients, aged five to 39 years of age, with mild lung disease who received oral ibuprofen or placebo twice daily for four years in doses to achieve peak plasma concentrations of 50 to 100 micrograms per ml. The patients on ibuprofen had a slower annual rate of change in FEV1 than the patients assigned to placebo (mean [+/- SE] slope, -2.17 +/- 0.57 percent vs. -3.60 +/- 0.55 percent in the placebo group; P = 0.02), and weight (as a percentage of ideal body weight) was better maintained in the former group (P = 0.02). Among the patients who took ibuprofen for four years and had at least a 70 percent rate of compliance, the annual rate of change in FEV1 was even slower (-1.48 +/- 0.69 percent vs. -3.57 +/- 0.65 percent in the placebo group, P = 0.03), and this group of patients also had a significantly slower rate of decline in forced vital capacity, the percentage of ideal body weight, and the chest-radiograph score. There was no significant difference between the ibuprofen and placebo groups in the frequency of hospitalization. One patient was withdrawn from the study because of conjunctivitis, and one because of epistaxis related to ibuprofen.
The authors concluded that in patients with CF and “mild” lung disease, high-dose ibuprofen, taken consistently for four years, significantly slows the progression of the lung disease without serious adverse effects. However, despite these positve results, the benefits of ibuprofen in this trial were not convincing to most clinicians. Subsequently the modest effect and frequent side effects were reasons there was not more widespread use of ibuprofen and the treatment never became popular even in the United States – by 2012 only some 3.3% of patients 6-12 yrs old on the CF Foundation’s registry were taking the drug.
For example Fennell PB et al (J Cyst Fibros 2007; 6:153-158.[PubMed]) reported half their patients stopped ibuprofen because of side effects (mainly gastrointestinal pain and bleeding) and the treatment had no effect on either the rate of pulmonary decline or hospitalisation rates in those who tolerated the drug. However, subsequent reports from Michael Konstan remained supportive (Konstan MW et al, Am J Resp Crit Care Med 2007; 176:1084-1089.[PubMed]) and Larry Land’s separate Canadian study showed slower FVC but not FEV1 decline (Lands LC et al. J Pediatr 2007; 151:249-254.[PubMed]).
An interesting and unrelated observational report suggested the recurrence of nasal polyps was reduced while patients were receiving ibuprofen (Lindstrom DR et al. J Otolaryngol 2007; 36:309-314. [PubMed]).
Professor Michael Konstan (figure 40) is Director of the Leroy Matthews Cystic fibrosis Centre, at the Rainbow Babies and Childrens Hospital, Cleveland and particularly identified as being associated with this study. He is a leading figure involved in CF care and research in the USA and heavily involved in both.
1995 Khan TZ, Wagener JS, Boast T, Martinez J, Accurso FJ, Riches DWH. Early pulmonary inflammation in infants with cystic fibrosis. Am J Respir Crit Care Med 1995; 151:1075-1082.[PubMed]

Fig 41. Jeffrey Wagner
This paper from Denver is frequently quoted as providing evidence of the presence of inflammation in the airways in the absence of infection. Bronchoalveolar lavage fluid (BALF) from 16 infants with CF and 11 disease control infants was examined for a variety of inflammatory parameters. Each index of airway inflammation was increased in the BALF of infants with CF as compared with control infants – including those with negative microbiological cultures for common CF-related pathogens, common respiratory viruses and fungi at the time of bronchoalveolar lavage (BAL).
The authors concluded that these findings suggested that airway inflammation was already present in infants with CF as young as four weeks. However the authors did not know if the seven infants with evidence of inflammation but negative cultures had received treatment previously for infections from their own paediatricians prior to being referred to them for bronchoscopy i.e. was there residual inflammation from a previous treated bacterial infection?
The number with positive lower respiratory tract cultures was high (9/17) although the care of some infants was with the local referring paediatrician not the authors. Although, from other publications, it seems very likely that there is a tendency to an excessive inflammatory response in the CF airways, it is doubtful if this study established that non-bacterial inflammation is common in young screened CF infants. For example the study of Armstrong et al 1996 (below) shows cells and inflammatory markers only where there was also bacterial infection.
It is therefore likely that inflammation does not occur without infection but when it does occur the degree of inflammation tends to be excessive and prolonged.
Dr Jeffrey Scott Wagner (fig. 41) of the University of Colorado Children’s Hospital is heavily involved in both CF care and research in the USA. He is a Board member of the Colorado CF Fondation, involved in various national trials and advisory committees.
1995 Littlewood JM. Abdominal pain in cystic fibrosis. J R Soc Med 1995; 88 (Suppl): 9-17.[PubMed]
A detailed review of the causes, investigation and treatment of abdominal pain in CF summarising experience with over 600 patients with CF seen at the Leeds Regional Paediatric CF Centre. As sub-specialisation increased in paediatrics in the UK, the majority of respiratory paediatricians working in CF centres were primarily interested in the treatment of the chest – understandably as most people with CF died from this cause. So the gastrointestinal aspects were relatively under-investigated although a significant number of patients had, and still do have, abdominal symptoms.
In this group of patients recurrent abdominal pain occurred in 11% of children receiving their care at the Leeds CF centre (where there was considerable interest in and investigation of gastrointestinal problems) and in 31% of those referred to Leeds for Comprehensive CF Assessment from other hospitals – admittedly a selected group.
It was interesting that the prevalence of recurrent abdominal pain differed significantly with the mode of presentation e.g. meconium ileus 38%, respiratory 39%, malabsorption 47%, presence of a CF sibling 30% but only in 4% in those diagnosed by neonatal screening and treated from the age of a few weeks (Littlewood JM J R Soc Med 1992; 85 (Suppl 18):13-19.[PubMed]).
1995 Ghosal S, Taylor CJ, Pickering M, McGraw J, Beckles-Wilson, Wales JKH. Disproportionate head growth retardation in cystic fibrosis. Arch Dis Child 1995; 72:150-152.[PubMed]
Fifty children with CF (18 diagnosed with meconium ileus, four by postnatal screening, 30 by clinical diagnosis) were followed over four years. The length SD scores improved from -1.24 at birth to -0.15 at four years and the weight SD scores from -1.37 at 6 months to -0.53 at four years. In contrast the head circumference SD score reached a plateau of -1.0 from the age of 1.5 to four years and remained significantly low throughout the four years of measurement being -1.05 at four years. Also there is evidence that malnutrition in infancy can affect intellectual development in the first five years (Lloyd-Still JD et al, Pediatrics 1974; 54:306-311.[PubMed]).

Fig 42. Shamik Ghosal
This was the first report that the head circumference of children with CF may be slightly smaller than expected. The high proportion of infants with meconium ileus (34%) in the series was almost certainly due to Sheffield Children’s Hospital being a regional centre for neonatal surgery. The authors suggested that the data may support the expression of CFTR in the choroid plexus and ependyma.
There is very little in the literature on head circumference in CF although in the Wisconsin screening data on cognitive score index (CSI) – “the highest proportion of CSI scores >84 occurred in the control <300E group (41%). “Patients in this group also had the lowest mean head circumference z-scores at diagnosis” (Koscik RL et al. J Pediatr 2005; 147(3 Suppl):S51-6. [PubMed]).
A further study of screened CF infants from Sheffield confirmed that head growth appeared to lag behind somatic growth supporting the functional expression of CFTR in the brain (Ghosal S et al. Arch Dis Child 1996; 75:191-193.[PubMed]
Dr Shomik Ghosal (fig. 42) was working at the the Sheffield Children’s Hospital at the time of this study and subsequently became consultant paediatric gastroenterologist at Staffordshire Hospital.
1995 Morkeberg JC, Edmund C, Prause JU, Lanng S, Koch C, Michaelsen KF. Ocular findings in cystic fibrosis patients receiving vitamin A supplementation. Graefe’s Arch Clin Exp Ophthalmol 1995; 233:709-713.[PubMed]
Only 26% of 35 patients examined in the Copenhagen clinic had normal vitamin A status as measured by serum retinol and light sensitivity but reduced contrast sensitivity. Conjunctival imprints showed dry eye in 42%; decreased tear stability in 49% and other abnormalities of low tear production (31%) and increased numbers of dying cells (23%). In fact 26% were considered to have the criteria for “keratoconjunctivitis sica”. The authors even suggested that the high incidence of dry eye could be a primary manifestation of CF.
These findings are more likely the result of suboptimal vitamin A status particularly as only 26% had normal vitamin A levels. In studies where vitamin A status is regularly monitored to maintain normal serum levels, such as Leeds, only reduced contrast sensitivity is found (Ansari et al. 1999 below) and the cause of this is unexplained.
1995 Dankert-Roelse JE, Meerman GJ. Long term prognosis of patients with cystic fibrosis in relation to early detection by neonatal screening and treatment in a cystic fibrosis centre. Thorax 1995; 50:712-718.[PubMed]
Fig 43. Jeanette Dankert-Roelse. Author’s photo
Comparative clinical follow up in three birth cohorts of patients with CF was performed at the Cystic Fibrosis Centre in Groningen. The first birth cohort (n = 19) was detected by screening and the two other cohorts were detected clinically, one (n = 30) consisting of patients born during the screening programme and the other (n = 32) of patients born during the six years immediately after the screening programme ended.
Patients born during the screening programme but detected clinically appeared to have a reduced life expectancy compared with patients detected by screening. The patients detected by screening showed less deterioration in lung function, a smaller increase in immunoglobulin levels, and minimal catch-up growth compared with the non-screened birth cohort of the same age. Expert management when started immediately after an early diagnosis of CF by neonatal screening results in important beneficial effects on the outcome and clinical course of the condition. The institution of very early treatment may be critical for the outcome and long term prognosis for most patients with cystic fibrosis. The authors believe that neonatal screening programmes for cystic fibrosis should be introduced more widely.
Jeanette Dankert-Roelse (fig. 43) from the Netherlands has been a tireless advocate of CF neonatal screening since the early Eighties and is still, in 2014, involved in the European CF Society neonatal CF screening programme. It is a relief that the obvious benefits of neonatal screening are now accepted and supported by studies that are acceptable to virtually all. However, it was 10 years before neonatal CF screening was routine in the Netherlands.
1996 Armstrong, DS. Grimwood K, Carlin JB, Carzino R, Olinsky A, Phelan PD. Bronchoalveolar lavage or oropharyngeal cultures to identify lower respiratory pathogens in infants with cystic fibrosis. Pediatr Pulmonol 1996; 21:267-275.[PubMed]
A study designed to determine whether oropharyngeal cultures predicted the presence of pathogens in the lower airways. In children with CF during 1992-1994, 75 of 90 (83%) infants with CF diagnosed by neonatal screening had 150 simultaneous bronchoalveolar lavage (BAL) and oropharyngeal specimens collected for quantitative bacterial culture at a mean age of 17 months (range, 1-52 months). Ten children undergoing bronchoscopy for stridor served as controls. Some, in fact many, of the infants with CF were currently receiving antibiotics – either anti-Staphylococcal antibiotics (44) or inhaled tobramycin (11). Total and differential white cell counts and interleukin-8 concentrations were measured in BAL fluid. A subset of bacterial pathogens was typed by pulsed field gel electrophoresis. A non-linear relationship with inflammatory markers supported a diagnosis of lower airway infection when > or = 10(5) colony-forming units/ml were detected. This criterion was met in 47 (31%) BAL cultures from 37 (49%) children. Staphylococcus aureus (19%), Pseudomonas aeruginosa (11%), and Hemophilus influenzae (8%) were the major lower airway pathogens. In oropharyngeal cultures, S. aureus (47%), Escherichia coli (23%), H. influenzae (15%), and P. aeruginosa (13%) predominated. The sensitivity, specificity, and positive and negative predictive values of oropharyngeal cultures for pathogens causing lower respiratory infections were 82%, 83%, 41%, and 97%, respectively. When there was agreement between paired oropharyngeal and BAL cultures, genetic fingerprinting showed some strains of the same organism were unrelated.
The authors concluded that oropharyngeal cultures do not reliably predict the presence of bacterial pathogens in the lower airways of young CF children.
This is an important study if only for showing that an alarming number of infants with CF had infected lower airways even at an average age of only 17 months. In many clinics, such as Copenhagen, children with CF have frequent cultures, every month or more often – and whenever they are unwell. The multiple cultures then available allow a more accurate assessment of the likelihood of lower respiratory tract infection over time – rather than the “one off” correlation as occurred in this and a number of similar studies. Also this practice of evaluating frequent throat cultures as an indicator of lower respiratory infection is supported by Pseudomonas antibody levels which correlate with the culture results.
So if the upper respiratory cultures are repeatedly negative it is likely that the lower airways are also uninfected – such an infant will virtually always have negative Pseudomonas antibody levels.
1996 Doull IJ, Freezer NJ, Holgate ST. Growth of prepubertal children with mild asthma treated with inhaled beclomethasone dipropionate. Am J Resp Crit Care Med 1996; 151:1715-1719. [PubMed]
The effect of 7 months inhaled beclomethasone propionate 400 micrograms/day on linear growth and adrenal function in 94 children aged seven to nine years. Mean regressed daily growth was significantly decreased during the treatment period 0.79 mm versus 1.14 mm per week. At the end of the seven months study the BDP treated children had grown significantly less than the children on placebo (mean 2.66 versus 3.64 mm). There was no catch up over 4 months.
So inhaled BDP at these relatively modest doses in children with mild asthma significantly decreased statural growth – a side effect so vigorously denied for so long by many leading respiratory paediatricians since we first reported this side effect (Littlewood et al, 1988 above). This is important in the context of CF as many children are treated, at times unnecessarily, with inhaled steroids (Balfour-Lynn I M et al. Am J Resp Crit Care Med 2006; 173:1356-1362. below), The doses used may be large and growth impairment is not a rare finding.
1996 Carles S, Desgeorges M, Goldman A, Thiart R, Guittard C, Kitazos CA, de Ravel TJ, Westwood AT, Claustres M, Ramsay M. First report of CFTR mutations in black cystic fibrosis patients of southern African origin. J Med Genet 1996; 33:802-804. [PubMed]

Fig 44. Michelle Ramsay
Cystic fibrosis was thought to be rare in the black populations of Africa and only a few patients have been reported but they had not been studied at the molecular level. This report, from Michelle Ramsay’s laboratory, concerns the detection of CFTR mutations in three black South African patients. One was homozygous for the 3120 + 1G–>A mutation, while the other two were compound heterozygotes each with this mutation on one chromosome. The other mutations were G1249E and a previously unreported in frame 54 bp deletions within exon 17a involving nucleotides 3196-3249 (3196del54).
The 3120 + 1G–>A mutation was first described in black American patients and has been shown to be a relatively common mutation in this population (9-14% of CF chromosomes). It was also found in a black CF patient whose father, the 3120 + 1G–>A carrier, is from Cameroon. These data suggest that it is an old mutation which accounts for many of the CFTR mutations in black Africans (Also Pileggi A. 1962; Kulczycki LL et al, 1964 above and other reports find a low incidence in black patients).
Prof. Michelle Ramsay (fig. 44) is head of the Molecular Genetics Laboratory in the Division of Human Genetics and the National Health Laboratory Service and the University of Witwatersrand.
1996 Brock DJH. Prenatal screening for cystic fibrosis: 5 years’ experience reviewed. Lancet 1996; 347:148-150.[PubMed]

Fig 45. David Brock
Antenatal screening had been available at two maternity clinics in Edinburgh, UK, since January, 1992, first on a research basis and then routinely. 25,000 couples had been screened. The take-up rates for the two-step and couple models of delivery were very similar at about 70%. Of 22 high-risk couples identified entirely through screening, 20 (91%) opted for prenatal diagnosis. Four couples returned for second and two for third monitored pregnancies.
In all eight cases where affected fetuses were identified, pregnancy was terminated. David Brock concluded that “these data remove one of the few remaining obstacles to a general implementation of prenatal screening for CF”.
Although prenatal screening was recommended in the UK by a Health Technology Assessment (Murray et al, 1999) and after this was accepted in principle by the National Screening Committee, prenatal screening had not been introduced in the UK by 2023. Furthermore, antenatal CF screening was discontinued in the Edinburgh hospitals in 2005 apparently on grounds of both cost and also the introduction of neonatal screening and the evidence of improving prognosis for infants with CF diagnosed soon after birth (also Mennie et al, 1992 above; Livingstone et al, 1994 above).
1996 Bikangaga P, Canny GJ. Benign intracranial hypertension in infants with cystic fibrosis. Arch Pediatr Adolesc Med 1996; 150:551-552. [PubMed]
Four of 53 (7.7%) newly diagnosed infants with CF in Toronto developed transient benign raised intracranial pressure during their initial treatment.
This is an apparently benign occurrence according to previous reports (Roach ES, Sinai SH. Clin Pediatr 1989; 27:371-373.[PubMed]). Vitamin A deficiency has been described as a cause (Abernathy RS. AJDC 1976; 130:1360-1362 above) also systemic steroid withdrawal. Catch up growth after severe malnutrition has also been implicated but many cases are unexplained. The complication has also been described with tetracycline antibiotics since the Sixties.
1996 Cheng K, Smyth RL, Govan JRW, Doherty C, Winstanley C, Denning N, Heaf DP, Saene H van, Hart CA. Spread of ß-lactam resistant Pseudomonas aeruginosa in a cystic fibrosis clinic. Lancet 1996; 348:639-642.[PubMed]

Fig 46. David Heaf
A high proportion of children attending the Liverpool Paediatric CF centre were found to be chronically infected with a P. aeruginosa that was resistant to ceftazidime and other beta-lactam antibiotics. Two genomic fingerprinting techniques were used to see if this had arisen from epidemic spread of a single strain. This had indeed occurred and 92 (76.7%) of the 120 children attending the clinic were infected with P aeruginosa, and 65 (71%) of these 92 infected infants harboured isolates that were resistant to ceftazidime; 55 of the 65 children harboured the same epidemic strain – resistant to ceftazidime, azlocillin, and imipenem, and sensitive to tobramycin and ciprofloxacin.
This study provides the first molecular evidence of a long-term “outbreak” of P. aeruginosa in a CF centre. The authors suggested that careful surveillance of the prevalence of antibiotic resistance in CF centres should be instituted with measures to prevent cross-infection. They suggested that anti-Pseudomonal monotherapy “should be considered with caution”.
This lesson had already been learned in Copenhagen in the early Eighties (Pedersen et al, 1986 above) and in the past a number of writers had already cautioned against the use of monotherapy. Most CF centres at the time already followed the recommendation to use two antibiotics when treating exacerbations of Pseudomonas infection. However, prior to this outbreak, intravenous ceftazidime monotherapy had been routine in the Liverpool CF clinic.
This was a very important paper which highlighted the risk of cross infection with Pseudomonas aeruginosa in CF clinics now clearly identified by genomic finger printing techniques. Later this highly transmissible “Liverpool epidemic strain” of Pseudomonas aeruginosa was reported elsewhere and subsequently spread to many other CF centres in the UK.
It is cause for concern that, even after experience such as reported here, there is still discussion as to the use of one or two antibiotics for the treatment of exacerbations – although it is true that a Cochrane review failed to give firm advice to use two antibiotics!! (Ephick HE, Tan A. Single versus combination intravenous antibiotic therapy for people with cystic fibrosis. Cochrane Database of Systematic Reviews. 2005).
Dr David Heaf (fig. 46) was the Director and senior consultant paediatrician at the Alder Hey Children’s Hospital CF Unit in Liverpool. He and his Liverpool colleagues developed an extensive shared care service for children with CF in the North West of England and North Wales.
1996 Frederiksen B, Lanng S, Koch C, Hoiby N. Improved survival in the Danish center-treated cystic fibrosis patients: results of aggressive treatment. Pediatr Pulmonol 1996; 21:153-158. [PubMed]

Fig 47. Christian Koch, Birgette Frederikson, Niels Hoiby, Jim Littlewood. Copenhagen 1997.
Survival data for Danish center-treated CF patients, covering the period 1974-1993. The annual mortality rate for 1989-1993 was 0-1.2%. Using the age-specific mortality rate for 1989-1993, it was impossible to calculate the median survival probability because the curve did not fall below 50% (age up to 45 years); however, it was possible to show that the survival probability for a newborn CF child to reach his 45th birthday was 80.4% (confidence interval 76.5-84.6%). The median age at diagnosis was 0.63 years with no sex difference. Surprisingly neonatal CF screening was not introduced in Denmark until 2016. The probability of surviving 40 years after the diagnosis of CF was made was 83.3% (confidence interval 80.1-86.6%).
These survival figures are considerably better than any other published survival probability at the time. The only item where I would disagree with the Danish CF centre is their policy of not introducing neonatal screening until 2016 (Marianne Skov et al, 2020 pubmed.ncbi.nlm.nih.gov/31682332/)
Dr Birgitte Frederiksen (fig. 47) was a senior clinician at the Copenhagen CF Centre and has been involved in and coordinated many of the studies from that unit.
1996 Robinson M, Regnis JA, Bailey DL, King M, Bautovich GJ, Bye PTP. Effect of hypertonic saline, amiloride, and cough on mucociliary clearance in patients with cystic fibrosis. Am J Respir Crit Care Med 1996; 153:1503-1509.[PubMed]
After inhalation of hypertonic (7%) saline alone, and with amiloride, the amount of radio aerosol cleared from the right lung at 60 and 90 minutes was significantly increased. The authors suggested that inhaled hypertonic saline was a potentially useful treatment for CF.
Peter Bye’s colleagues at the Adult CF Centre at St Vincent’s Hospital in Sydney continued their work on hypertonic saline and eventually carried out a successful clinical trial of hypertonic saline in adults with CF and confirmed the value of hypertonic saline treatment (Elkins et al, 2006 below).
Hypertonic 7% saline inhalations were originally reported to double the rate of removal of bronchial secretions in chronic bronchitis in an interesting study using radio aerosols (Pavia Det al, Am Rev Respir Dis 1978; 117:199-203.[PubMed]). I recall consulting Margaret Hodson in the early Eighties asking her for any suggestions for treating a girl with CF who had excessively viscid secretions. She advised a trial of nebulised hypertonic saline which, from her personal experience, had proved helpful!!
1997 Katznelson D, Szeinberg A, Augarten A, Yahav Y. The critical first six months in cystic fibrosis: a syndrome of severe bronchiolitis. Pediatr Pulmonol 1997; 24:134-136.[PubMed]

Fig 48. Daniel Katznelson. Author’s photo
The syndrome of infantile bronchiolitis in cystic fibrosis carries a high mortality. Fifteen cases of CF encountered over the past 19 years with severe bronchiolitis with onset during the first 6 months of life are described. Treatment includes steroids in high doses. All patients recovered. Further progress resembled the usual natural course of CF and showed no evidence of persisting lung damage. The mechanism of this syndrome is not clear and is probably dependent on many factors involved in early lung disease in CF. The frequency of severe bronchiolitis in cystic fibrosis may not be high, but it continues to be seen in clinical practice today.
This paper is one of many on CF and a wide range of paediatric subjects by Professor Daniel Katznelson of the National CF Center, Sheba Medical Center, Israel.(fig. 48)
1996 Colombo C, Battezzi PM, Podda M, Bettinadi N, Giunta A. Ursodeoxycholic acid for liver disease associated with cystic fibrosis: a double blind multicenter trial. Hepatology 1996; 23:1484-1490. [PubMed]
A trial of ursodeoxycholic acid by Carla Colombo and colleagues from Milan who first reported the use of URSO in people with CF (Colombo et al, 1990 above). Fifty five patients from 12 CF centres over one year had either UDCA + taurine, UDCA + placebo, placebo + taurine or double placebo. The UDCA treated patients showed better clinical condition and improved biochemistry; also those treated with taurine had improved prealbumin levels.
Despite this trial, a Cochrane review, revised in 2009, considered “There is insufficient evidence to justify its (URSO) routine use in cystic fibrosis”. The most recent Cochrane review states “There are few trials assessing the effectiveness of ursodeoxycholic acid. The quality of the evidence identified ranged from low to very low. There is currently insufficient evidence to justify its routine use in cystic fibrosis”.
The latest publication on this subject in 2023 – J Cyst Fibros . 2022 Mar;21(2):220-226. doi: 10.1016/j.jcf.2021.03.014.Epub 2021 Apr 2. pubmed.ncbi.nlm.nih.gov/33814323/
Conclusions: CF patients followed-up in UDCA prescribing centers did not show a lower incidence of portal hypertension as compared to those followed in centers not prescribing UDCA. These results question the utility of UDCA in reducing the occurrence of severe liver disease in CF.
However, some clinicians would use URSO at an early stage if there is any evidence of liver involvement.
1997 Pollitt RJ, Dalton A, Evans S, Hughes HN, Curtis D. Neonatal screening for cystic fibrosis in the Trent region (UK): two-stage immunoreactive trypsin screening compared with a three-stage protocol with DNA analysis as an intermediate step. J Med Screen 1997; 4:23 [PubMed]

Fig 49. Rodney Pollitt
To assess neonatal screening for cystic fibrosis using immunoreactive trypsin, either alone or in conjunction with DNA analysis for the delta F508 mutation. A novel three-stage screening protocol was compared with the previously introduced two-stage immunoreactive trypsin-DNA protocol.
(a) Collection of data from a 4 1/2 year period (phase 1) of two-stage immunoreactive trypsin screening. The initial dried blood samples were obtained at 6 days of age and repeat samples at 27 days of age from babies with results above the 99.5th centile. Babies with persistent hypertrypsinaemia were referred for a diagnostic sweat test. (b) Retrospective DNA analysis: patients with cystic fibrosis diagnosed in phase 1 were genotyped and most samples from babies with increased initial immunoreactive trypsin but normal results in the second sample were analysed for the delta F508 mutation. (c) Phase 2, a prospective study of a three-stage neonatal screening protocol, in which only babies heterozygous for the delta F508 cystic fibrosis mutation progressed to the second immunoreactive trypsin test
437 859 babies born between August 1989 and March 1996. main outcome was proportions of unaffected babies requiring a second blood sample or a sweat test. Overall sensitivity for the detection of cystic fibrosis.
The two-stage screen failed to identify six out of 94 cases of cystic fibrosis (without meconium ileus). The introduction of the DNA analysis step would have resulted in one additional case being missed. With the three-stage screen there was a 92% reduction in babies requiring a second blood sample and an 80% reduction in negative sweat tests, results close to the predictions of the retrospective study.
The authors concluded that the three-stage screening protocol is a marked improvement on the two-stage immunoreactive trypsin strategy and on the two-stage immunoreactive trypsin-DNA strategy recently introduced in some other screening programmes.
Rodney Pollitt (fig. 49) is Honorary Professor, Division of Child Health, University of Sheffield, Consultant in Neonatal Screening, Sheffield Children’s Hospital and had a major influence on the introduction of national Neonatal CF screening in the UK.
For over 30 years Dr. Pollitt has been professionally involved in newborn screening locally and nationally. During this time he has become a true newborn screening guru locally, nationally and internationally. This is evidenced by his large number of publications (more than 150 book chapters and full papers in addition to a large number of conference abstracts) on newborn screening and metabolic disease topics. In 2002 he was awarded the Robert Guthrie Award of the International Society for Neonatal Screening
1997 Conway SP, Pond MN, Watson A, Etherington C, Robey HL, Goldman MH. Intravenous colistin sulphomethate in acute respiratory exacerbations in adult patients with cystic fibrosis. Thorax 1997; 52:987-993.[PubMed]
Intravenous colistin was shown to be an effective safe treatment for P. aeruginosa associated pulmonary exacerbations in patients with cystic fibrosis. Assessment of the individual effect of each treatment regimen suggests a greater efficacy when colistin is combined with a second antibiotic to which the Pseudomonas shows in vitro sensitivity. It was advised that renal function should be monitored.

Fig 50. Martin Goldman
Intravenous colomycin was first used by Robert Stern who gives an interesting account of his decision to use the intravenous route, instead of the painful intramuscular route, in a wasted girl who already had an intravenous drip running for hydration purposes. Colomycin was the only parenteral antibiotic in the early Sixties. Later an intravenous preparation became available and but was soon replaced by intravenous gentamicin and then carbenicillin and piperacillin became available. Stern describes the early developments including the heparin lock in the early Seventies, the gradual involvement of the patients with CF in managing their own IV therapy first in hospital and eventually at home (Stern RC. Intravenous treatment: where we are and how we got there. In: Doershuk CF, editor. Cystic Fibrosis in the Twentieth Century. Cleveland: AM Publishing, Ltd, 2001:93-111)
Dr Martin Goldman (figure 50) is an independent consultant Pharmaceutical physician with very broad hands on experience. He was responsible for taking Colobreathe from concept to authorisation when Senior Medical Advisor with Forest Laboratories (1989-2013). He has been closely associated with CF progress for many years.
1997 Everard ML, Sly P, Brenan S, Ryan G. Macrolide antibiotics in diffuse panbronchiolitis and in cystic fibrosis. Eur Respir J 1997; 10:2926. [PubMed]

Fig 51. Mark Everard
Professor Mark Everard (fig. 51) confirmed with me that this was the first report from Sheffield UK and Perth Western Australia showing the anti-inflammatory effect of macrolides in people with cystic fibrosis.
Four of six patients with CF had significant reduction in IL-8 sputum levels after one month treatment with low dose (200 mg tds) oral erythromycin. He believes the first report of the use of macrolides in CF was in a Japanese publication by Nakanishi et al in 1995 – “A 16-year-old boy was admitted to our hospital because of coughing, sputum, and exertional dyspnoea. Seven months after birth cystic fibrosis had been diagnosed. The chest roentgenogram on admission showed diffuse reticulonodular shadows and overinflation. Pulmonary function tests revealed obstructive and restrictive impairment. Erythromycin and Lomefloxacin were administered by mouth, and aminoglycosides were administered by inhalation. His symptoms were alleviated, and he is now an outpatient. In Japan, cystic fibrosis is rare, and this patient is extremely rare because he has grown up to be a 16-year-old. In this case, low-dose and long-term erythromycin administration was very effective”. (Nakanishi N, Ueda N, Kitade M, Moritaka T. A case of cystic fibrosis in a Japanese student. Jpn J Thoracic Dis 1995; 33:771-774. [PubMed]
1998 Jaffe A, Francis J, Rosenthal M, Bush A. Long term azithromycin may improve lung function in children with cystic fibrosis. Lancet 1998; 351:420.

Fig 52. Adam Jaffe
In a short report in 1998 Adam Jaffe and colleagues at the Royal Brompton showed an impressive improvement in seven severely affected children with CF. Daily azithromycin for more than 3 months increased FEV1 and FVC by more than 11%. The beneficial effect of 600 mg/day of erythromycin for 1 to 12 months in chronic panbronchiolitis in Japanese patients had been reported previously (Nagai H et al. Respiration 1991; 58:145-149. [PubMed] The effect appeared to be independent of the presence of chronic Pseudomonas infection and an anti-inflammatory action was suggested (Fujii T et al. Thorax 1995; 50:1246-52.[PubMed]; Hoiby N. Thorax 1994; 49:531-532.[PubMed]; Kudoh S et al. Jpn J Thorac Dis 1987; 25:632-42.[PubMed]; Kudoh S. et al. Am J Respir Crit Care Med 1998; 157:1829-32. [PubMed] below).
These were important developments eventually leading to a number of large clinical trials and the widespread use of azithromycin in people with CF – undoubtedly one of the major clinical treatment advances of the Nineties and new Millennium (Equi et al, 2002 below; Saiman et al, 2003 below). By 2010 70% of all the patients over 6 years old on the CF Foundation Registry were taking long term azithromycin.
Adam Jaffé (fig.52) eventually became a Paediatric Respiratory Consultant at Sydney Children’s Hospital and the John Beveridge Professor and Head of Paediatrics at the University of New South Wales.
1997 Wallis C, Leung T, Cubbitt D, Reynolds A. Stool elastase as a diagnostic test for pancreatic function in children with cystic fibrosis. Lancet 1997; 350:1001.[PubMed]

Fig 53. Colin Wallis
The first published report of faecal pancreatic elastase 1 in children with CF from Colin Wallis, (fig. 53) paediatrician at Great Ormond Street, London. All non-CF children had normal faecal elastase levels but 26 of 30 children with CF had undetectable levels. Three of the others were pancreatic sufficient. Normal levels were reached by normal infants within 2 weeks.
The test was further evaluated in a larger series of children with CF in Leeds (Cade et al, 2000 below) and became the accepted method of determining pancreatic function. The test had a major advantage over faecal chymotrypsin as it still remained low (negative) in pancreatic insufficient patients whilst the patient was taking pancreatic enzyme supplements.
This test was a major advance – a really useful standardised non-invasive test for accurately confirming the presence of pancreatic insufficiency without having to stop the pancreatic enzyme treatment of patients referred to a CF Centre for full investigation – a far cry from duodenal intubation for tryptic activity which was essential before the sweat test was introduced during the Fifties; also it was a major advance on faecal chymotrypsin. However, faecal chymotrypsin did provide a useful indication as to whether the patient was taking their pancreatic enzyme supplements – a low chymotrypsin when thought to be taking pancreatic enzymes suggested non-adherence.
1997 Button BM, Heine RG, Catto-Smith AG, Phelan PD, Olinsky A. Postural drainage and gastro-oesophageal reflux in infants with cystic fibrosis. Arch Dis Child 1997; 76:148-150. [PubMed]

Fig 54. Brenda Button. Author’s photo
First of series of papers from Brenda Button (fig. 54), a physiotherapist from Melbourne, Australia, noting the possible dangers of inhalation when infants with CF were in the head down position during postural drainage; infants with CF were known to have an increased incidence of gastro-oesophageal (GO) reflux. Physiotherapy with and without head down tilt were compared using 24 hour pH oesophageal monitoring. Standard physiotherapy with head down tilt was associated with a significant increase in GO reflux in the infants with CF.
This was an important study and, with Brenda Button’s subsequent publications, certainly had a major influence on the techniques of physiotherapy recommended for CF infants (see also Malfroot & Dab, 1991 above for earlier studies on reflux in CF infants; Button et al, 2004 also confirmed GO reflux as common and important in adults with CF). Later studies from the Brompton Hospital in London questioned the findings of reflux in the head down position (Phillips GE et al. Holding the baby: head downwards positioning for physiotherapy does not cause gastro oesophageal reflux. Eur Resp J 1998; 12:954-957. [PubMed]
1997 Drittanti L, Masciovecchio MV, Gabbarini J, Vega M. Cystic fibrosis: gene therapy or preventive gene transfer? Gene Therapy 1997; 4:1001-1003.[PubMed]
I was very impressed by this paper, which few clinicians have read, for it accurately described the situation seen by those who follow many patients year after year. Patients’ respiratory function tests are usually stable before chronic infection (usually Pseudomonas) becomes established but begin a slow but relentless deterioration after chronic Pseudomonas infection becomes established i.e. they pass the “point of no return” (figure 55 – my figure)..
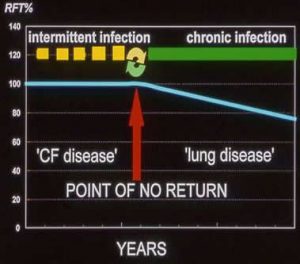
Fig 55. The “point of no return” This author’s illustration
These authors suggested that before the onset of chronic infection people with CF can be considered to be in the phase of “CF Disease” i.e. they have various physicochemical alterations in the electrolyte and liquid composition of the airway fluid but no tissue damage. Although they get respiratory infections these can be eradicated, if treated early with antibiotics and their respiratory function remains stable.
However, eventually a new respiratory infection is not eradicated and chronic infection of the bronchial and pulmonary tissues becomes established. At this stage the patient then enters the phase of “Lung disease” where infection and chronic inflammation become established, self perpetuating and progress independently of the basic CFTR abnormality – this change is referred to as the“Point of no return” and there follows a slow deterioration in the patient’s condition, the speed of which is determined by the type and intensity of treatment givenn receive.
So the aim of modern treatment is to avoid passing the “Point of no return” i.e. prevent or delay for as long as possible the onset of chronic infection of the airways. So it can be appreciated that if the airway cultures are repeatedly positive for Pseudomonas, even if the patient has few or even no symptoms (as in the paper by Konstan et al, above), he/she already has passed the “Point of No Return”.
In a later paper Drittanti is one of the authors who state that “The interactivity between the CFTR gene and cystic fibrosis would be limited to the initial phase of the disease” (Genetics in Medicine 2000;2:124-130.[PubMed]). While the initial phase was related to the CFTR genotype, the kinetics of the second phase seems to be common to all groups considered. The hypothesis that the interactivity between the CFTR gene and CF disease would be limited in time is presented, suggesting that mutant CFTR would trigger a disease that evolves to become independent from the CFTR gene itself.
1997 Frederiksen B, Koch C, Hoiby N. Antibiotic treatment at time of initial colonisation with Pseudomonas aeruginosa postpones chronic infection and prevents deterioration of pulmonary function in patients with cystic fibrosis. Pediatr Pulmonol 1997; 23:330-335.1[PubMed]

Fig. 55a Bernadette Frederriksen
Follow up of the important Valerius et al, 1991 study (above) by Bernadette Frederiksen (Fig.55a) showing successful eradication of early infection with P. aeruginosa with nebulised colistin and oral ciprofloxacin. Three months colistin and ciprofloxacin gave longer freedom from recurrence of infection than did three weeks treatment.
This study was criticised as historic controls were used. However, it was already quite obvious that the early eradication treatment prevented chronic P. aeruginosa infection (Littlewood et al, 1985; Valerius et al, 1991 above). Vigorous early eradication treatment was already routine practice in many European CF clinics and most clinicians would have regarded as unethical the involvement of a placebo group at this stage. It is difficult to understand how three months treatment increases the time to further Pseudomonas infection as the next episode of infection is usually, but not always, with a different genotype presumably acquired from the environment (Munck A et al. Pediatr Pulmonol 2001; 32:288-292. [PubMed]). However, also using genotyping, it was later shown that a minority of Pseudomonas strains were suppressed but not completely eradicated – so presumably three months treatment would reduce the chances of this occurring.
The great delay in the general introduction of early eradication therapy for Pseudomonas to reduce the prevalence of chronic infection, particularly in North America, was difficult to understand when the results of the earlier Copenhagen study were so impressive and so different from the expected (Valerius et al, 1991 above).
Surprisingly, a later Cochrane Systematic review on early eradication of Pseudomonas aeruginosa, almost a decade later in 2006, grudgingly could only conclude that “there is some evidence that antibiotic treatment of early P. aeruginosa results in short term eradication but it remains uncertain whether there is clinical benefit to people with cystic fibrosis” . The most recent Cochrane review (2 June 2023) states “There is insufficient evidence to determine whether these antibiotic strategies decrease mortality or morbidity, improve quality of life, or are associated with adverse effects compared to placebo or standard treatment” – a conclusion most experienced clinicians, including Niels Hoiby would fiercely question (as he and I did in critical letters to the earlier Cochrane reviewers!). The views of the late Christian Koch summarise the Danish experience and of most experienced CF clinicians as to the importance of avoiding chronic Pseudomonas infection (these are quoted in the comments on Valerius et al, 1991 above).
1997 McIlwaine PM, Wong LT, Peacock D, Davidson AG. Long-term comparative trial of conventional postural drainage and percussion versus positive expiratory pressure physiotherapy in the treatment of cystic fibrosis. J Pediatr 1997; 131:506-508. [PubMed]

Fig 56. Maggie Mcllwaine. Author’s photo
Forty patients were randomised to receive either postural drainage and percussion or PEP mask physiotherapy over one year. Those using the PEP mask had significantly better respiratory function after 1 year and the authors concluded that this was the more effective method of physiotherapy.
This study from Professor George Davidson’s CF centre in Vancouver, Canada had a significant influence on physiotherapy practice in North America. A subsequent study from this unit, comparing PEP and Flutter methods, showed PEP to be superior in maintaining pulmonary function and reducing the need for hospital admissions (McIlwaine et al. J Pediatr 2001; 138:845-850).
The study was influential in changing the routine physical therapy practice in North America.
Maggie Mcllwaine (fig. 56) is the Senior Physiotherapist at the British Columbia Children’s Hospital, Vancouver CF Centre and has made major important contributions to CF care.
1998 Lindblad A, Glaumann H, Strandvik B. A two-year prospective study of the effect of ursodeoxycholic acid on urinary bile acid excretion and liver morphology in cystic fibrosis-associated liver disease. Hepatology. 1998; 27:166-74. [PubMed]

Fig 57. Anders Lindblad
The efficacy of 2 years of treatment with ursodeoxycholic acid (UDCA) in cystic fibrosis (CF)-associated liver disease was evaluated by liver biopsies and liver function tests in 10 patients aged 8 to 28 years. Blind evaluation of liver biopsies indicated improved liver morphology with less inflammation and/or bile duct proliferation than before treatment with UDCA in 7 patients. Only 1 patient had signs of progression of clinical liver disease. The secondary bile acids, such as lithocholic acid (LCA) and deoxycholic acid (DCA), did not increase significantly. The excretion pattern of glycosidic conjugates of UDCA and its metabolites was similar to that found in healthy individuals, UDCA and isoUDCA being mainly excreted in conjugation with N-acetylglucosamine.
The authors conclude this study shows that UDCA modulates inflammation in CF-associated liver disease and indicates improvement of liver morphology during 2 years of treatment.
Dr Anders Lindblad (figure 57) of the Department of Pediatrics, Sahlgrenka University Hospital and the CF Centre, The Queen Silvia Children’s Hospital, Goteberg, Sweden.
1998 Ledson MJ, Tran J, Walshaw MJ. Prevalence and mechanisms of gastro-oesophageal reflux in adult cystic fibrosis patients. J R Soc Med 1998; 91:7-9. [PubMed]

Fig 58. Martin Walshaw
Fifty adults with CF were served by questionnaire and ten with reflux symptoms had oesophageal manometryand 24hr pH recordings,Forty seven patients (94%) had upper gastrointestinal symptoms: 40 (80%) heartburn (27 worse when supine); 52%) regurgitation; and 28 (56%) dyspepsia. At oesophageal manometry, lower oesophageal sphincter barrier pressure (LOSBP) was (subnormal in 6 of the 10 patients and 3 had uncoordinated peristalsis in the mid-oesophagus. Eight patients had significant gastroesophageal reflux. Adult CF patients have high rates of GOR symptoms, diminished LOSBP, and acid reflux.

Fig 59. Malcolm Ledson
This study re-emphasised the fact that GOR was a frequent and important problem and a significant cause of symptoms in adults with cystic fibrosis. The complication had been recognised since first described by Jan Feigelson (Feigelson & Sauvegrain, 1975 above) and later by Scott RB et al, 1985 (above); the frequency and importance was again highlighted in this present paper. Brenda Button in Australia had already pointed out the problem of the occurrence of reflux in CF infants in the head down position during physiotherapy (Button et al, 1997 above) – later she also found reflux to be common in adults with CF (Button et al, 2004).
Dr Martin Walshaw (fig. 58) was the first Director of the Liverpool CF Centre for adults and was later joined by Dr Malcolm Ledson (fig. 59). The Liverpool CF Centre for adults is now one of the major units in the UK.
1998 Kudoh S. Azuma A. Yamamoto M. Izumi T. Ando M. Improvement of survival in patients with diffuse panbronchiolitis treated with low-dose erythromycin. Am J Respir Crit Care Med 1998; 157:1829-32.[PubMed]
Diffuse panbronchiolitis (DPB) is a chronic inflammatory disease of the airways with a high mortality despite treatment with a combination of antibiotics and the use of supportive therapy. Low-dose erythromycin therapy (EM) (400 to 600 mg/d) improved the survival and most patients in Japan have been treated with this regimen since 1984. The authors compared the survival rates of 498 patients with DPB after dividing them into three groups (Group a: 1970-1979, Group b: 1980-1984, Group c: 1985-1990). The survival rate of Group c was significantly higher than that of Groups a (p < 0.0001) and b (p < 0. 0001). In Group c (1985-1990), eight of 87 patients died; five (21%) died in the EM non-treated subgroup (n = 24), and three (5%) died in the EM-treated subgroup (n = 63).
So treatment with erythromycin was associated with a significant improvement in the survival of Japanese patients with DPB which was more significant in the older than in the younger patients. These findings were the basis for the gradual evaluation of macrolide therapy in people with cystic fibrosis. The first report of the favourable effect of erythromycin in DPD was Kudoh S, et al. Clinical effects of low-dose long-term erythromycin chemotherapy on diffuse panbronchiolitis. Jpn J Thorac Dis 1987; 25:632-642.[PubMed]). Mark Everard et al, (1997 above) was the first in the UK to report an effect of macrolides on sputum inflammatory markers in cystic fibrosis.
1998 Aris RM, Renner JB, Winders AD, Buell HE, Riggs DB, Lester GE, Ontjes DA. Increased rate of fractures and severe kyphosis: sequelae of living into adulthood with cystic fibrosis. Ann Int Med 1998; 128:186-193.[PubMed]

Fig 60 Robert Aris
Bone mineral density was measured with dual-energy x-ray absorptiometry, patient-reported fracture events were confirmed by radiography, and kyphosis angles were measured by using the Cobb method. Mean bone mineral densities for the spine, femur, and total body were severely depressed in patients with cystic fibrosis, averaging 2 SDs below those of age-matched normal controls (P<0.001). Patient interviews showed that 54 fractures had occurred over 1410 patient-years, and chest radiographs showed evidence of 14 additional rib and 62 additional vertebral compression fractures. The database (which covered 1410 patient-years) showed that fracture rates were approximately twofold greater in women with cystic fibrosis aged 16 to 34 years (P = 0.015) and men with cystic fibrosis aged 25 to 45 years (P = 0.04) than in the general population. Vertebral compression and rib fractures were 100- and 10-fold more common than expected, respectively (P<0.001 for both comparisons). The mean kyphosis angle (+/- SD) for this group was markedly abnormal (44 +/- 14 degrees; 62% > or = 40 degrees) and probably contributed to diminished stature (mean height loss, 5.8 cm in men with CF and 5.9 cm in women with cystic fibrosis). Cumulative prednisone dose, body mass index, and age at puberty were the strongest predictors of bone mineral density.
Although osteoporosis had been mentioned in a number of previous reports,(Gibbens DT et al. J Pediatr 1988; 113:295-300.[PubMed]), this was the first major study on osteoporosis in adults with cystic fibrosis. The study drew attention to the fact that osteoporosis is almost universal in adults with late-stage CF and its complications include increased fracture rates and severe kyphosis. The findings stimulated a great deal of research into osteoporosis in adults with cystic fibrosis.
Later the subject was reviewed in many publications and in a consensus document from the UK Cystic Fibrosis Trust (Bone Mineralisation in Cystic Fibrosis. UK Cystic Fibrosis Trust Bone Mineralisation Working Group. February 2007. Full text available on the UK CF Trust website (www.cftrust.org.uk)
Dr Robert Aris (fig. 60) is a member of the Pulmonary Diseases and Critical Care Medicine Department in the University of North Carolina. He is involved with the care of cystic fibrosis and has a major interest in osteoporosis on which he has published widely and has also contributed to both N. American and European Guidelines.
1998 Lowden J, Goodchild MC, Ryley HC, Doull I. Maintenance of growth in cystic fibrosis despite reduction in pancreatic enzyme supplementation. Arch Dis Child 1998; 78:377-378.[PubMed]

Fig 61. Iolo Doull
This study from Cardiff, in the post-fibrosing colonopathy era, further supported the view that children with CF in the UK were taking unnecessarily large doses of pancreatic enzyme.
Fifteen children with CF on a mean enzyme intake equivalent to 18,300 U lipase/kg/day were able to reduce their enzyme supplements to a mean of 8,647 U lipase/kg/day. There were no changes in energy or fat intake but significant increases in weight, height and weight for height.
Many UK patients were taking considerably more enzymes supplements than the equivalent of 10,000 U lipase/kg/day advised as a result of the occurrence of fibrosing colonopathy (Mehta A. Lancet 2001;358:1547-1548). This study from Cardiff supported this and confirmed that in some patients the large doses were not required.
Dr Iolo Doull (fig. 61) followed Mary Goodchild as the Director of the Cardiff Paediatric CF Centre. He is heavily involved in both CF care and research. He has developed a shared care network for most of Wales which involves him and his team travelling periodically to the various general hospitals in Wales. In 2012 he reported better respiratory outcomes for children receiving all their CF care at the main Cardiff CF Centre (Doull I et al. Arch Dis Child 2012; 97:17-20.[PubMed] below).
1998 Jaffe A, Francis J, Rosenthal M, Bush A. Long-term azithromycin may improve lung function in children with cystic fibrosis. Lancet 1998; 351:420. [PubMed]
The second very convincing observational study on macrolides and CF by Adam Jaffe (first report by Everard et al, 1997 above) showing very impressive improvement in the respiratory function of severely affected children given regular azithromycin in addition to their usual treatment. These impressive observations were followed by a controlled trial from the Brompton Hospital, London which confirmed the beneficial effect (Equi et al, 2002 below) and also by a major trial from the US CF Foundation (Saiman et al, 2003 below).
Dr Adam Jaffe (fig. 52 above) moved from London to the full-time position of Respiratory Specialist in Sydney Children’s Hospital Respiratory Department. He had worked in Sydney some 10 years previously at the Royal Alexandra Children’s Hospital in Camperdown (now known as The Children’s Hospital at Westmead) also in London as Head of the Academic Respiratory Medicine Unit at Great Ormond Street Children’s Hospital.
1998 Conway SP. Transition from paediatric to adult-orientated care for adolescents with cystic fibrosis. Disability & Rehabilitation 1998; 20:209-216.

61a. Steve Conway
The Leeds CF Centre had been running a monthly transition clinic for the previous 10 years since the adult CF unit was started, first at Seacroft Hospital in 1988; Dr Conway (fig.61a) had been involved since its inception. All patients should have the opportunity to transfer to a properly equipped and properly staffed adult cystic fibrosis centre where they can continue to receive the highest standards of care from an experienced multidisciplinary team.
In some cities the paediatric and adult CF units are closely related both geographically and administratively. For example, in Leeds Dr Conway, before he retired in 2013, was the “Lead Clinician” in both the paediatric and adult CF units. Also in Copenhagen the same team care for both children and adults. Certainly this arrangement does reduce the stress experienced by many patients (and their parents) in transferring to a new hospital and different CF staff. On the other hand some of the problems of small children and adults with CF are very different. Perhaps an ideal is to have a lead clinician involved with both units sharing the responsibility of the adults with an adult physician and of the children with a paediatrician. In Copenhagen it was eventually necessary to have a separate adult CF unit.
1998 Cunningham S, Marshall T. Influence of five years of antenatal screening on the paediatric cystic fibrosis population in one region. Arch Dis Child 1998; 78:345-348.[PubMed]
The incidence of CF in the five years before and after antenatal screening was introduced in Edinburgh decreased from 4.6 to 1.6 infants per year – a reduction greater than could be accounted for by prenatal diagnosis and termination.
Much of the early work on antenatal screening during the Eighties was done in Edinburgh by David Brock and his colleagues (Brock 1992; Livingstone et al. 1994; Brock, 1996 – all above). It is disappointing that the antenatal screening which Brock pioneered was eventually abandoned in Edinburgh and has not been introduced elsewhere in the UK. The introduction of neonatal CF screening in Scotland and the steady improvement in prognosis, being two reasons given for withdrawal of the antenatal screening in Edinburgh. Financial reasons prevented the introduction of antenatal screening in England even by 2023.
1998 Mahadeva R, Webb K, Westerbeek RC, Carroll NR, Dodd ME, Bilton D. Clinical outcome in relation to care in centres specialising on cystic fibrosis: cross sectional study. BMJ 1998; 316:1771-1775. [PubMed]

Fig 62. Ravi Mahadeva
This is one of the few papers which is accepted as supporting the superiority of CF Centre care to the care received at a general paediatric clinic in the local hospital. Patients at the adult cystic fibrosis centre were subdivided into three groups. Those who had received continuous care from paediatric and adult cystic fibrosis centres (group A), those who had received paediatric care at their local hospital then at an adult CF centre (group B) and those who had received neither paediatric nor adult specialist care (Group C). Body mass index was 21.3, 20.2 and 18.3 for Groups A B and C respectively (P<0.001) and the improved nutritional status was correlated with a higher FEV1 and better chest X-rays (P<0.001 for both).
These findings are widely quoted as providing the first direct evidence that management in cystic fibrosis centres resulted in a better clinical outcome. This had been appreciated for years by doctors dealing with people with CF, and indeed by patients and parents, but is still contested by a minority of general paediatricians and physicians.
Dr Ravi Mahadeva (fig. 62) is now a consultant Chest Physician in Cambridge UK. He has a long standing interest in mechanisms of pulmonary inflammation in COPD, particularly in those with alpha-1 antitrypsin deficiency.

Fig 63. Kevin Webb. Author’s photo
Professor Kevin Webb (fig. 63) started the first CF centre for adults in Manchester at Monsall Hospital in 1982; the unit later moved to the purpose-built Bradbury Unit at Wythenshawe Hospital in 1994. The CF Centre is one of the largest in the UK and Kevin and his colleagues have made major contributions to improving CF care. In 2013 Kevin received the ECFS Award “to recognise and honour the huge contribution his scientific research has had on our understanding of clinical care, genetics, and therapy – and the importance of cross infection of Burkholderia cepacia complex organisms and Pseudomonas aeruginosa for his contribution to CF”.
1998 Rosenstein BJ, Cutting GR for the Cystic Fibrosis Consensus Panel. The diagnosis of cystic fibrosis: a consensus statement. J Pediatr 1998; 132:589-595. [PubMed]
A helpful publication summarising the criteria for diagnosis as follows – One or more of the characteristic phenotypic features OR a history of CF in a sibling OR a positive neonatal screening result AND an increase in sweat chloride concentration (> 60mmol/l) OR identification of two CF mutations OR demonstration of abnormal nasal epithelial transport.

Fig 64. Beryl Rosenstein (R) Richard Grand (L) & Jim Littlewood at NACFC 2008
Dr. Beryl Rosenstein (fig. 64) is a Professor in the Department of Pediatrics, Johns Hopkins. He attended Boston University and received his medical degree from Tufts University. He completed his residency in pediatrics at Johns Hopkins, and has served as Director of Ambulatory Services in the Pediatric Emergency Department, Director of the Johns Hopkins Hospital Full-Term Nursery, Medical Director of the Mt. Washington Pediatric Hospital, and as Director of the Johns Hopkins Cystic Fibrosis Clinic. He was Vice President for Medical Affairs at the Johns Hopkins Hospital from 1994 to 2009. He has published many papers on a wide variety of aspects of CF and received a prestigious award from the CF Foundation for his work with CF patients.
Dr Richard Grand (fig. 64) is Professor of Pediatrics at Boston Children’s Hospital. He received his MD from New York University School of Medicine. He completed an internship and residency in pediatrics at Boston Children’s Hospital and fellowships at Massachusetts General Hospital, Massachusetts Institute of Technology and the National Institutes of Health. His clinical research has focused on growth and nutrition in chronic disease, with a particular emphasis on inflammatory bowel disease (IBD), His group was the first to recognize the nutritional basis of growth failure in IBD and to conceptualize a strategy for its therapy. Dr Grand is a member of the National Board of Directors, the Crohn’s and Colitis Foundation of America. He is the recipient of numerous honors and awards including the Harry Shwachman Award, North American Society for Pediatric Gastroenterology and Nutrition. His first paper on CF was in 1966 (Grand RJ et al. Pregnancy in cystic fibrosis. JAMA 1966; 195:993-1000) and more recently he was involved in the investigations into fibrosing colonopathy.
1999 Waters DL, Wilcken B, Irwing L, Van Asperen P, Mellis C, Simpson JM, Brown J, Gaskin KJ. Clinical outcomes of newborn screening for cystic fibrosis. Arch Dis Child 1999; 80: F1-F7.[PubMed]
An important long term observational study from Sydney of 10 year follow-up of screened infants with CF, showing significant nutritional and respiratory benefits from neonatal CF screening first introduced in 1981 in New South Wales. Fifty seven unscreened CF children (born 1978-1981) are compared with 60 screened infants (born 1981-1984). Height and weight of screened are consistently better (SDs of 0.3 and 0.4 better respectively) and respiratory function is higher – FEV1 difference of 9.4% and FVC 8.4% in favour of the screened infants. In New South Wales a more progressive attitude to neonatal CF screening has prevailed over the years since the early Eighties than in the UK.
Although Cochrane reviewers considered this paper unsatisfactory as the controls were historical, most people accepted that the results were predictable and provided further supportive evidence for neonatal CF screening.
1999 Ansari EA, Sahni K, Etherington C, Morton A, Conway SP, Moya E, Littlewood JM. Ocular signs and symptoms and vitamin A status in patients with cystic fibrosis treated with daily vitamin A supplements. Br J Ophthalmol 1999; 83:688-691. [PubMed]
Since the first Leeds studies on vitamin levels started in 1978 (Congden et al, 1981 above), oral supplementation has been guided by annual monitoring of vitamin levels estimated at Annual Assessments which have been routine since 1980, Also all patients are seen regularly by a specialist CF dietitian and most have a comprehensive annual dietary assessment. None of the 28 patients in this study had vitamin A deficiency, the median value of serum retinol being 48 microg/dl, range 31-80 microg/dl (normal range 30-80 microg/dl). Dark adaptation was normal in all patients compared with the control group where the mean value was 3.4 log units of threshold luminance (95% confidence interval 2.4-4.0). None of the test group had a value of threshold luminance 2 SD above the mean value for the control group. Eight patients had reduced contrast sensitivity. The median value for serum zinc was 14.2 micromol/ l, range 13-81 micromol/l (normal range 8-23 micromol/l) and the median value for retinol binding protein was 36 mg/l, range 13-81 mg/l (normal range 35-58 mg/l).
There was no correlation between dark adaptation and serum retinol, zinc, or retinol binding protein. Two patients had clinical evidence of dry eye.
So regular estimates of plasma vitamin A, together with appropriate supplementation according to plasma levels and expert dietetic review, can maintain normal dark adaptation in patients with cystic fibrosis. The occurrence of reduced contrast sensitivity function is well documented in CF and confirmed in this study but remains an unexplained phenomenon (also Morkeberg et al, 1995 above).
1999 Ramsey BW, Pepe MS, Quan JM, Otto KL, Montgomery AB, Williams-Warren J, Vasiljev-K M, Borowitz D, Bowman CM, Marshall BC, Marshall S, Smith AL. Intermittent administration of inhaled tobramycin in patients with cystic fibrosis. Cystic Fibrosis Inhaled Tobramycin Study Group. N Engl J Med 1999; 340:23-30.[PubMed]

Fig 65. Bonnie Ramsey
One of the most important clinical trials of the decade showing a significant benefit of inhaled preservative free tobramycin (TOBI) given during alternate four week cycles for 24 weeks to patients chronically infected with Pseudomonas aeruginosa. Treated patients had an average increase of FEV1 of 10% predicted at 20 weeks where as those on placebo had a 2% decline; also the treated patients had 23% fewer hospital admissions.
The introduction of TOBI, supported by this excellent clinical trial, was one of the major clinical advances of the decade and the culmination of work started on tobramycin in the late Eighties by Arnold Smith and others (Smith AL et al. 1989 above). By 2012 65.7% of people with CF aged at least 6 years and with chronic P. aeruginosa infection in the US CFF Registry were receiving nebulised TOBI.
Although the cost (£10K per annum in the UK) has restricted the use of the TOBI preparation in the UK, inhaled anti-Pseudomonal antibiotics (colistin, tobramycin for injection and gentamicin) have been widely used in the UK for CF patients with chronic Pseudomonas infection since Margaret Hodson’s important 1981 paper (Hodson et al, 1981 above).
Dr Bruce Montgomery (Figure 54) now Senior Vice President and Head of Respiratory Therapeutics at Gilead Sciences, was closely involved with the development and trials of this preparation and has been involved subsequently with other new treatments including, more recently, nebulised Cayston (aztreonam lysine for inhalation). In 2010 Bruce received the Distinguished Corporate Award of the CF Foundation for his work in drug development for people with CF.
1999 Haworth CS, Selby PL, Webb AK, Dodd AK, Musson H, McL Niven R, Economu G, Horrocks AW, Freemont AJ, Mawer EB, Adams EJ. Low bone density in adults with cystic fibrosis. Thorax 1999; 54:961-967. [PubMed]

Fig 66. Charlie Howarth
An early detailed study of a population of 151 adults aged 15-52 years with CF from Manchester using DEXA and quantitative computed tomography and biochemical markers of bone turnover.
34% of adults with CF had BMD Z scores of -2 or less at one or more skeletal sites. The respiratory function and physical activity were related to the BMD Z scores. Markers of bone turnover were negatively related and vitamin D positively related to the BMD Z scores despite supplementation with vitamin D.
Dr Charlie Haworth (figure 66) has made major contributions to the increasingly important bone problems in CF both when working in Manchester and after moving to Papworth CF Adult centre where he is now Director.
Subsequent reviews and consensus statements on bone problems in CF followed from North America and the UK
2005 Aris RM, Merkel PA, Bachrach LK, Borowitz DS, Boyle MP, Elkin S, Guise TA, Hardin DS, Haworth CS, Hollick MF, JosephM, O’Brien K, Tullis E, Watts NB, White TB. Consensus statement: Guide to bone health and disease in cystic fibrosis. J Clin Endocrinol Metab 2005; 90:1888-1896. [PubMed]
2007 Bone mineralisation in cystic fibrosis. Report of the UK Cystic Fibrosis Trust Bone Mineralisation Working Group. London. Cystic Fibrosis Trust, February 2007. Conway S (Chairman), Compston J, Cunliffe H, Dodd M, Elkin S, Haworth C, Jaffe A, Morton A, Redfern J, Truscott J. (full text on CF Trust website www.cftrust.org.uk) (figure 56). Last updated 2013.
1999 Vavrova V, Zemkova D, Bartsova J, Zapletal A, Smolikova L. Krebsova A, Macek M jr. Cystic fibrosis – a disease of adolescents and adults. [Czech]. Casopis Lekaru Ceskych 1999; 138:654-659.[PubMed]

Fig 67. Vera Vavrova receiving the ECFS Award from the President, Stuart Elborn in 2008
The summary of this article gives an account of the situation regarding CF in the CF centre of the Faculty Hospital in Prague Motol in 1999. 349 patients are followed; also included are the 95 who died since 1985. 126 (36.1%) survived to the age of 18 years. The median age of death increased from 12.2. years in 1985-1990 to 18.8 years in 1991-1998. Adult status was satisfactory 40.4%, poor in 33.3% and marginal in 26.3%. Pulmonary function was normal in 17.5%, and severely affected in 22.8% – the rest between 40-80% predicted. Modern intensive treatment has improved the prognosis and quality of life of people with CF.
Professor Vavrova (figure 67) of Prague published her first paper on CF in 1962 and her most recent in 2009. She has been involved in CF care and research for nearly 50 years and involved in over 80 papers dealing with many aspects of the condition. She received the ECFS Award in 2008.
1999 Sokol RJ, Durie PR. recommendations for the management of liver and biliary tract disease in cystic fibrosis. Cystic Fibrosis Foundation hepatobiliary Disease Consensus Group. J Pediatr Gastroenterol Nutr 1999; 28 Suppl 1:S1-13. [PubMed]
Recommendations of an expert group on management of CF related liver disease.
1999 Connett GJ, Lucas JS, Atchley JT, Fairhurst JJ, Rolles CJ. Colonic wall thickening is related to age and not dose of high strength pancreatic microspheres in children with cystic fibrosis. Eur J Gastro Hepatol 1999; 11:181-183. [PubMed]

Fig 68. Garry Connett
Thirty three children with CF are described, including 25 who had been receiving high strength pancreatin in the form of Creon 25,000 continuously for 3 years. Median lipase intake was 19,330 u/kg/day (range 0-59,000). There was no relationship between enzyme dosage and colonic wall thickness. The most important relationship of colonic wall thickness appeared to be with age.
This was a very useful report and reassuring that high doses of pancreatic enzymes, when given in the form of Creon 25,000

Fig 69. Chris Rolles
, did not appear to cause colonic damage. It had already been evident from the UK CF Database that many UK patients were taking more than the recommended equivalent of 10,000 U lipase/kg/day (Mehta A. Lancet 2001; 358:1547-1548). Obviously it would be unwise to translate these findings to other brands of high strength enzymes.
Dr Garry Connett (figure 68) is a Paediatric Respiratory Consultant who succeeded Dr Chris Rolles as Director of the Southampton Paediatric CF Centre. He is heavily involved in CF care and clinical research.
Dr Chris Rolles (figure 69) is President of Child Health International and previously was consultant paediatrician at Southampton where he founded the Paediatric CF Centre. Chris has done a great deal to improve the care of people with CF in a number of countries including Russia.