
Some previous publications on the history of cystic fibrosis mentioned in this section
1972 Cystic Fibrosis. A Comprehensive Bibliography of the Medical Literature 1813-1972. US Department of Health E1ducation and Welfare. National Institutes of Health. Complied and edited by Douglas S Holsclaw and Anne Lloyd Topham.
1981 1000 years of Cystic Fibrosis. Collected Papers. Presented at an International Conference on Cystic Fibrosis at the University of Minnesota, May, 1981. Warren J Warwick Ed. Published by the University of Minnesota.
1984 HISTORY OF THE CYSTIC FIBROSIS (RESEARCH) TRUST A lecture by Sir Robert Johnson QC delivered at the CF Research Trust’s 20th Anniversary Weekend, Brighton, June 1984
1989 Dodge JA. Cystic fibrosis in Wales. Welsh Paed J 1989; 1:4-6. (www.welshpaediatrics.org.uk/cystic) Full text on the web.
1990 Kulczycki LL. Five decades of cystic fibrosis (1938-1988). Acta Universitas Carolinae Medica 1990; 36:7-12.
1992 Super M. Milestones in cystic fibrosis. In; Warner JO, editor. Cystic Fibrosis. British Medical Bulletin 1992; 4:717-737.
1993 Mearns MB. Cystic Fibrosis: the First 50 Years. A review of the clinical problems and their management. In: Dodge JA, Brock DJH, Widdicombe JH, editors. Cystic Fibrosis – Current topics. Vol I. Chichester: John Wiley and Sons, 1993: 217-250.
1994 Dietzsch H J Research and treatment in cystic fibrosis (in honour of the work of Harry Shwachman). Eur J Pediatr. 1994 Nov; 153(11): 783. https://pubmed.ncbi.nlm.nih.gov/7843189/
1998 Welsh MJ, Ramsey BW. Research on cystic fibrosis: a journey from the Heart House. Am J Respir CritCare Med 1998; 157 Suppl: S148-S154.[PubMed]
1999 Quinton PM. Physiological basis of cystic fibrosis: a historical perspective. Physiol Rev 1999; 79: S3-S22 [PubMed]
2001 Cystic Fibrosis in the 20th Century. People Events and Progress. Doershuk CF (Ed). A M Publishing Ltd, Cleveland, Ohio. 2001
2004 Wellcome Witnesses to Twentieth Century Medicine. Volume 20. Cystic Fibrosis. Christie DA, Tansey EM. (Eds). Wellcome Trust Centre for the History of Medicine at University College. London.
2004 The Joseph Levy Memorial Lecture. “Looking back over 40 years and what the future holds”. Littlewood J M
2008 Fanos JH. “We kept our promises”: An oral history of Harry Shwachman, MD. Am J Med Genet Part A 146A:284-293.[PubMed]
2014 Stephanie Clague. Dorothy Hansine Andersen. Lancet Respir Med 2014 Mar;2(3): 184-185. https://pubmed.ncbi.nlm.nih.gov/24621679/
2016 Wilmott Robert W. An invited article. “A Historical Perspective of Cystic Fibrosis”. Hong Kong Journal Paediatrics 2016; 21:280-285.
2016 Hodson and Geddes’ Cystic Fibrosis. Fourth Edition Eds. Andrew Bush, Diana Bilton & Margaret Hodson. CRC Press. Taylor and Francis Group.
1971 Busch R. Zur m der Mukoviscidose beim Neugenboren. (Comprehension of mucoviscidosis in the newborn). Kinderarztl Prax 1971; 39:268-271. (German)[PubMed]
1978-2005 Further articles by Busch R with brief abstracts
1971 Busch R. Zur m der Mukoviscidose beim Neugenboren. (Comprehension of mucoviscidosis in the newborn). Kinderarztl Prax 1971; 39:268-271. (German)[PubMed]007
2007 Westwood T, Ramsay M. Medical/Scientific topic. Something New Out of Africa: Cystic Fibrosis Affects All Peoples. http://www.cfww.org. CF Worldwide Newsletter Jan 15th 2007.
2007 “History of the European Working Group for Cystic Fibrosis & European Cystic Fibrosis Society Conferences” A lecture by Professor Niels Hoiby adapted by Dr Jim Littlewood
2009 Robert Steven Kaplan, Sophie Hood. “Bob Beall at the Cystic Fibrosis Foundation”. Published by The Harvard Business School. 9-409-107. April 17th 2009. Copies and permissions from www.hbsp.harvard.edu/education.
2013 Watt P. For the Roses. Cystic Fibrosis Ireland. Published by Cystic Fibrosis Ireland (www.cfireland.ie)
2014 Stephanie Clague. Dorothy Hansine Andersen Lancet Respiratory Medicine. Historical Profile Lancet Vol 2, Issue £, p184-185. 2014.
2018 Philip Farrell. Discovering the ancient origin of cystic fibrosis, the most common genetic disease in Caucasians. The Conversation. 2018 September 7 (below)
2018 Philip Farrell, Claude Férec, Milan Macek, Thomas Frischer, Sabine Renner, Katherina Riss, David barton, Teresa Repetto, maria Tzetis, Karine Giteau, Morton Duno Melissa Rogers, Hara Levy, Mourad Sahbatou, Yann Fichu, Cedreic Le Marechal , Emmanuelle Genin. Estimating the age of p.(Phe508del) with family studies of geographically distinct European populations and the early spread of cystic European Journal of Human Genetics volume 26, pages 1832–1839 (2018) Free PMC article pubmed.ncbi.nlm.nih.gov/30089827/
****************************************************
1972 Cystic Fibrosis. A Comprehensive Bibliography of the Medical Literature 1813-1972. US Department of Health Education and Welfare. National Institutes of Health. Complied and edited by Douglas S Holsclaw and Anne Lloyd Topham.


Fig 1. Douglas Holsclaw
Details of over 5000 references on CF published between 1813 and 1972.
This must have been a massive undertaking in the pre-computer and pre-internet era. The entries are in alphabetical order of first author and there are also useful separate subject and author indices.
By 2013 there were over 37,000 references to cystic fibrosis on the medical reference databases; by the end of 2019 the number of references to cystic fibrosis on the PubMed database has risen to 51,991.
I was fortunate to discuss this major undertaking with Douglas Holsclaw when I obtained Fig.1.
1981 1000 years of Cystic Fibrosis. Collected Papers. Presented at an International Conference on Cystic Fibrosis at the University of Minnesota, may 7-7-, 1981. Warren J Warwick Ed. Published by the University of Minnesota.

Fig 2. Warren Warwick
Warren Warwick (figure 2) organised this “one off” meeting where many of the leading international CF experts, past and present, presented a fascinating collection of papers on their work over the previous years. It is a wonderful collection of papers presented by virtually everyone with a significant involvement in CF treatment or research up to that time. Many had been involved with CF care or research since the first recognitioon of the condition in 1938 by Dorothy Andersen.
Presenters at the meeting included Warren Warwick, Guilo J Barbero, Paul di Sant’Agnese, Jack Lieberman, LeRoy Matthews, Lewis Gibson, Archie Norman, Lucas Kulczycki, Alexander Spock, Jean Feigelson, Jean Navarro, Niels Hoiby, Winge Flensborg, Peter Sciotz, Ettore Rossi, Richard Kraemer, Lynne Reid, KF Kerrebijn, Nancy Huang, Gianni Mastella, Ulrich Stephan, William Waring, George Polgar, Bettina Hillman, Carl Doershuk, Harry Shwachman.
============================================================
1984 HISTORY OF THE CYSTIC FIBROSIS (RESEARCH) TRUST
A lecture by Sir Robert Johnson QC delivered at the CF Research Trust’s 20th Anniversary Weekend, Brighton, June 1984

Fig 3. Sir Robert Johnson
Cystic fibrosis itself has, I suppose, a history as old as man. Until it’s eventual recognition by the medical world, fatalities from CF were attributed to other causes so that the number of people actually suffering from CF must be a matter of speculation. On the one hand, lack of diagnosis and therefore proper treatment must have meant that there were few living people with cystic fibrosis. On the other hand the number of carriers must have expanded unabated.
To put our Trust and the progress it has made into a historical perspective we must, I think, pause to remember that for many centuries cystic fibrosis existed but as a totally unrecognised condition. We all know of the Hungarian saying of many years ago – “soon will die the child whose brow is salty to the kiss” – I recollect seeing Ron’s (Ron Tucker) slide of that actually attributed to Rocholz, I think in 1857. The first report of cystic fibrosis in a scientific paper was in 1936 in the paper by Dr Guido Fanconi entitled, it may be noted Mr. Chairman, Cystic Fibrosis of the Pancreas. It is again a measure of that timescale in which I am recounting this history that is within my own, if not within your lifetime Mr Chairman. As that title implies the disorder was thought initially to relate only to the digestive system. The involvement of the lungs, to which our major problems seem now to relate, was not then recognised.

Fig 4. Dorothy Andersen
In 1938 there appeared the first comprehensive account of CF as a separate disorder based on work at the Babies Hospital at the Columbia Presbyterian Medical Center in New York City and written by Dr. Dorothy Andersen (figure 4), a name we all know, but significantly a pathologist. The following year there occurred the first diagnosis of a living CF patient. The diagnosis, obviously difficult for the doctor and uncomfortable for the patient, was achieved by the measurement of the pancreatic enzymes in the duodenal juice.
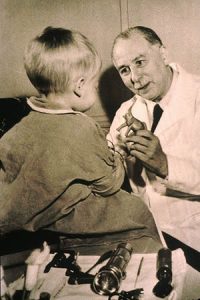
Fig 5. Sydney Farber
By 1944 however, it had become clear to the few doctors who had the knowledge, that CF was not confined only to the pancreas but was a widespread defect affecting other exocrine, that is to say secreting, glands. Cystic fibrosis of the pancreas was therefore a misnomer and a new name had to be invented. Dr Sydney Farber (figure 5), of Boston Massachusetts, named it Mucoviscidosis. All of us recognise in that word, the word mucus, but some of the more erudite will have recognised in it the Latin “viscidus” meaning sticky as well as the Greek “osis” meaning condition; condition of sticky mucus. To this day the International CF Association is called the ICFMA, the International Cystic Fibrosis (Mucoviscidosis) Association because that’s what cystic fibrosis is called in some other countries and you will find mucoviscidosis is defined still as Fibrocystic Disease of the Pancreas. It was then seen that the mucus of the respiratory tract was affected. The realisation roughly coincided fortunately with the development in the late 1940s of the early antibiotics which had evolved since the discovery of penicillin by Sir Alexander Fleming in 1929, working in that small second floor room in that gatehouse building at St Mary’s Hospital, Paddington where Professor Williamson is now carrying out important work for us. Fleming discovered penicillin but it wasn’t until 1941 that it became available for practical use.

Fig 6. Paul di Sant’Agnese
In 1953, taking my chronology a stage further, an important step forward occurred in relation to CF in the detection of the CF defect in the sweat gland. This discovery occurred following a heat wave in the United States when it was observed that CF children were suffering heat prostration due to heavy salt loss. The discovery was made by Dr. Paul di Sant’Agnese (figure 6) – a doctor still alive (in 1984) who one hopes we will be seeing at this Congress next week. That was at the Babies Hospital in New York, again. That important discovery led eventually to the development of the sweat test.
In 1956, in the United States, they created their Cystic Fibrosis Foundation which in it’s early years made research grants to researchers whose names are household names in the world of cystic fibrosis – Dr Harry Shwachman (figure 7), Dr Paul di Sant’Agnese and Dr Dorothy Andersen. In 1959, ladies and gentlemen, only in 1959 there was introduced the ionisation technique (the Gibson and Cooke pilocarpine iontophoresis method of sweat stimulation and collection) which forms the basis of the sweat test as we know it and a major tool in the diagnosis of cystic fibrosis.

Fig 7. Archie Norman
It is only now Mr Chairman, at this stage in my chronology, that I can usefully turn your attention to the evolution of CF work in the United Kingdom. The fact is that until then there had been little of a coordinated nature to merit inclusion in my history.True there were individual doctors, Dr Norman over there (figure 7) to name one as an example, who were all working in developing ways of managing the patients better, but the general picture here was of ignorance and distress unmitigated by hope or practical effective action.
Until 1964 there was no formal organisation in the United Kingdom of any kind for the support or coordination of CF research. Work was going on but it was sparse and those involved were isolated and facing the problems of any scientific pioneer. As to the organisation of parents there is only one of which I am aware. It was based in Somerset and consisted of a body of parents who had infrequent meetings and kept in touch by means of a duplicated newsletter. The meetings and the newsletter seemed to me to be largely devoted to the exchange of emotive and dismal family news, and in my view of it at the time, of little benefit to the members. Perhaps for this reason the group had little momentum and it suffered the additional handicap of being regarded by the medical profession as detrimental to the families involved. Certainly it gave families the feeling that they were not alone, but there was no hope and nothing practical.
At the time of the creation of our charity, I recall attempts to absorb the Somerset group in the Chest and Heart Association and to make that the main CF organisation in the United Kingdom, but fortunately that failed. I say fortunately because that charity would have been concerned with too wide a range of disabilities to give adequate emphasis to Cystic Fibrosis; I remember this was a great source of anxiety in the early years until the matter was resolved. What CF did not need was fragmentation amongst a number of competing groups.
The situation of young parents facing a diagnosis of CF and trying to adjust to its implications will never be easy. However, if one compares the position now with the position when our Trust was founded one has a useful measure of the value of the contribution which the Trust has made in this area. Diagnosis will probably always be occasionally missed or delayed. In those days, missed diagnosis and late diagnosis seemed to be the order of the day. Scarcely any general practitioners had knowledge of cystic fibrosis and such indeed, with respect to them, seemed to be the case even with some consultants. We cannot yet claim total elimination of professional ignorance but now it is very much the exception. The promotion that is necessary for fundraising has contributed to that in a way coupled with the present massive research effort which is often seen to be reflected in the medical press.

Fig 9. Percy Lovely

Fig 8. John Panchaud
For the parents too, there is now available a range of information in understandable form which explains the meaning and significance of the diagnosis. None of that, I can tell you from my own experience, was available in former days. Further more, nowadays at our Groups and Branches there is available, in a thoroughly responsible way, advice and guidance from other parents. The Executive Committee is at the moment embarked on a scheme for some formal training in this voluntary counselling which is such an important part of the work of the Group and Branch Officers. But to revert to my history the picture I want to give you of the situation of a parent in the 1950s and the early 1960s is one of total hopelessness and despair. There was however one parent who was not content to wait passively for the inevitable. That parent as you know was John Panchaud (figure 8). His daughter Caroline had been diagnosed as having cystic fibrosis. John Panchaud’s father-in-law was a man called Percy Lovely (figure 9), a man highly respected in the City of London and a member of the Common Council.
At this time the Chairman of the National Dock Labour Board was Lord Crook who lived in Carshalton. In 1963 Lord Crook and the National Dock Labour Board gave a cocktail party at the Board’s Headquarters on the Albert Embankment in London to which Lord Crook invited a friend and neighbour from Carshalton, someone called David Lawson.

Fig 11. David Lawson

Fig 10. Lord Crook
David Lawson (figure 11) was a paediatrician at Queen Mary’s Children’s Hospital in Carshalton and himself a CF parent. Among the other guests, at what was a City function for business dignitaries, were, by chance, Mr and Mrs Percy Lovely. In the course of the party Dr Lawson espied Mrs Lovely. She was apparently a lady of some elegance wearing on this occasion a particularly edible hat! Having attracted, so it seems, the attention of this young Dr Lawson, and finding that he was a doctor, she poured out her heart to him about the problem her grand-daughter Caroline. It was left that she would get her son-in-law John Panchaud to phone David Lawson to arrange a meeting. Mr. Chairman, it is in that cocktail party, in the unlikely setting of the National Dock Labour Board Headquarters in London, that one has the origin of the Cystic Fibrosis Research Trust.
The consequence was that John Panchaud went to see Dr Lawson and together they evolve a plan. David Lawson’s first suggestion was that they should set up a Medical or Scientific Committee. John Panchaud proposed this should include David Lawson and a physician who was looking after Caroline at Great Ormond Street – Dr Archie Norman. But who else?

Fig 12. Cedric Carter

Fig 13. Winifred Young
Eventually Dr Lawson became Chairman of the Steering Committee. It included Dr. Norman and the late Dr. Cedric Carter (figure 12) the famous geneticist, the late Dr. Winifred Young (figure 13) of the Queen Elizabeth Children’s Hospital in Hackney – one of the early pioneers in the care of CF children and Dr. Lynn Reid (figure 14) of the Brompton Hospita, now a professor in the United States. A short time later they were joined by Dr. John Batten another physician from the Brompton who, as you know, is now the head of the Medical Household of Her Majesty the Queen.
In 1963 John Panchaud had his solicitor draw up the necessary legal documents for the formal creation of the charity. One of the first Trustees was Mr. Percy Lovely and as a result of his and John Panchaud’s contacts in the City they got together a Board of Trustees including as originals Mr. Joseph Levy (figure 16)and Lord Crook. Others were Lord Bossom and other prominent figures in the City of London. The inaugural meeting was held in February 1964. I have here the minutes. The date is the 20th February 1964 at 3.30pm in the Mansion House in the City of London under the auspices of the then Lord Mayor, Sir James Harman.

Fig 15. John Batten

Fig 14. Lynne Reid
The Trust organisation in those days consisted of John Panchaud’s own secretary, Eileen Denham, who spent part of his and quite a bit of her own time in the affairs of the Trust. The premises were John Panchaud’s office in the Haymarket. The prime movers on the fund raising side were John Panchaud himself, Percy Lovely and Joseph Levy. They set about the seemingly impossible task of raising funds for researching into something of which scarcely anyone had heard. David Lawson and his committee set about the allocation of the funds and giving general advice. One of these early minutes that I’ve read records that because the Medical Steering Committee was putting forward such a wide ranging programme of research, it would be necessary for the Trustees to now raise £10,000 per year. One sees from that the measure of the progress we’ve made.
At first the organisation was limited to the raising of funds by the individual Trustees. However,it gradually became clear that parents had to be involved. John Panchaud had always been keen to enlist their help and I think perhaps the initial delay or inertia in

Fig 16 Joseph Levy
enlisting the help of parents came from the experience the doctors had had from parents groups. The doctors were reluctant to encourage the organisation of parents into what might prove, once again, to be little more than gatherings of the emotional, the bitter and frustrated. I think there was an increasing realisation that there would be parents groups and it was felt better to take the initiative and organise them in a proper way rather than leave them to developing in a random and negative fashion. I think too that it was realised that if research was to be effective the fundraising base of the organisation needed to be broadened and the hope was that this could be done in a way that was beneficial to the cause without detriment to the families.
Here the initiative was taken by Dr Norman. He selected one of the CF mothers (Linda Johnson – please see addendum) in his clinic at Great Ormond Street to organise the first group in London. I say in London but the existence of that first group acted as a magnet and parents wrote and telephoned and came to meetings from all over the United Kingdom. There was a small group at the time I recollect in Sheffield led by Mr Albert Clover and almost simultaneously there were groups starting at Aberystwyth – Mrs Jones I recollect and Dr Glencross in Glasgow.
Suddenly there was hope. Suddenly, there was something one could do to fight back. A chance even to win. I look back on those days with great emotion. For those of us who were involved those days still mark a turning pointing in our lives, when hope arose and work began. After a depressing history of chaos and despair one man had been instrumental in bringing together a group of people from totally diverse backgrounds and uniting them into what was truly a crusade. A crusade to do something about it. Of course each of those I have mentioned was a person of talent and dedication in their own field, but without the catalyst of John Panchaud, it is I think doubtful that we would be here today celebrating the 20th Anniversary of a lively and successful charity. There would have been an organisation but not one of this calibre.

The John Panchaud Medal of the CF Trust
Of our founders some are still with us, others have passed on. Lord Crook as you know is our President, 83, and because of ill health not able to be with us this week. Joseph Levy (figure 16) is the much respected, and I wish to say much loved, Chairman of our Council. Dr Norman is just coming to the end of his term as Chairman of the Scientific and Medical Advisory Committee. Mr Lovely has died and so has John Panchaud. He died in 1974 when he was 58. His memory lives on. We have inaugurated, as you know a John Panchaud Medal (figure) which is to be awarded on rare occasions to those who have given outstanding service to the Trust. John’s widow is I hope to be at this Congress. Caroline is married and living in Switzerland.
Dr Lawson continued for many years his enormous endeavour on the research side. He was, as you know, Chairman of our Medical Committee and again of the Medical and Scientific Committee of the international organisation and has been, in many ways, on the medical side one of the major figures in the International CF Organisation but most importantly, has come out of his supposed retirement to play a major part in the organisation of the Scientific Programme of this Conference. I myself was a minor figure on the Organising Committee and I bear witness to the enormous effort that David Lawson and every member of the Committee put into the congress that we hope is going to be a great success this week.
To return to my history, after the formation of the Trust and that first parents group, the enthusiasm and the pace was colossal. Judged by today’s figures the funds seem small but they were hard earned and they were well spent, and they primed the pump. As early as Christmas 1964, the Trust was selling its own Christmas cards. I remember a meeting at the house of Dr Smith near Hangar Lane in West London, to choose a range of cards. The meeting I recall was much too large for the purpose and there was no consensus but somehow cards were chosen and the name of Cystic Fibrosis began to appear on other peoples’ mantelpieces. Dr Smith himself is worth a mention. He worked I think for the Wellcome Foundation doing research into asthma. His CF daughter was a source of wonder to the rest of us. She was, believe it or not, taking O levels. At that time when the expectation of life of our children was 3 or 4 years, here was a sign of hope if ever there was one. When she went to university I remember thinking to myself that surely she couldn’t have Cystic Fibrosis at all. There must have been a misdiagnosis, but such, ladies and gentlemen, was the situation in which we parents were in those early days.

Fig 17. Ron Tucker
No history of this Trust, however brief, could be written without mention of the services of Ron Tucker (figure 17). He was born on the 29th June so his birthday, his 65th in fact, is in 3 weeks time, when he will retire. Ron had a proud war service, he was wounded at Dunkirk, he went with the Commandos on the Dieppe raid and he was mentioned in dispatches. Before the war and during the war he became interested in youth work and when he left the army became full time organiser of boys’ clubs in Lancashire and Cheshire. By 1964 he was in London as Organising Secretary of the Keystone Cops, an organisation made up of famous and influential people who raised money for boys clubs. In 1965 the time had already come when the Cystic Fibrosis Trust needed, although it could scarcely afford, a full time executive. Advertisements were placed and interviews arranged. An interviewing panel was selected of those whose judgement in these matters could, so it was hoped, be relied on to select an appropriate candidate. I suspect, although I am not sure, that it was because of Joe Levy’s support of the boys clubs movement, that Ron Tucker was asked to be one of the pa
The interviews were concluded, the panel was unanimous. There had been no suitable candidate. The whole operation it seemed had been in vain. As the meeting broke up in an atmosphere of some gloom and disappointment, a panel member, addressing himself to Ron Tucker, said “Why don’t you do it?” Whether the remark was a flippant attempt at a joke to dispel the gloom or was a serious enquiry, I cannot relate. However, it was in that casual remark that there came to the service of Cystic Fibrosis, the life of Ron Tucker, to whose dedication and enthusiasm this Congress and our presence here today is modest tribute. This is not the occasion for a definitive account of the contribution made by Ron Tucker. We all of us know the enormous expansion in our organisation, the number of branches formed, the money raised, the families helped, almost single-handedly by Ron Tucker.

Fig 18. HRH Princess Alexandra with HRH Prince Charles – the present Patron
I want too, in my history, to mention the contribution made by Her Royal Highness Princess Alexandra (figure 18) . She takes a genuine and helpful interest in our endeavours but, particularly for the purpose of my history, I want to remind you that she became our Patron very early in the life of this charity, in 1968 at a time when this charity was struggling for recognition. Her granting of her patronage to us gave us a status which helped us very considerably. (Prince Charles (figure 18) succeeded Princess Alexandra as Patron in 2014).
I have already mentioned the international body. There had been some initial earlier contact between doctors on an international basis initiated I think by Dr. Paul di Sant’Agnese, involving in the UK significantly Dr Norman and Dr Lawson who were recognised as leaders of the UK medical effort. The first Congress of the ICF(M)A was held in Paris in March 1965. The practice has since become to hold such Congresses every 4 years but there were four earlier ones in the USA which were closer together in time and the Paris meeting was called the fifth; the last one in 1980 was in Toronto and the one in Brighton this week is the Ninth.
This is not the first International Congress to be held in England. Again, it is a measure of the progress that was made and one must add the esteem in which our UK researchers were held by the international medical and scientific community. As early as 1969 the International Congress was held in England. The funds available to the executive committee, the resources available to it were limited. We were asked to underwrite a pretty substantial project in terms both of organisation and finance, but everyone recognised the enormous benefit that can flow from bringing together in one place CF researchers from all over the world. We took over one of the Cambridge colleges, Churchill College, so that the visiting scientists and doctors were all accommodated under one roof. The format of the conference has become the pattern for subsequent international medical and scientific gatherings. That conference proved, I think, to be one of the most successful of the international conferences. Let us hope that this Congress is another such success and that the information and ideas that are exchanged and talked about here will speed the ambition of us all to find improved techniques of clinical management and to identify the basic defect.
In this account of the history of the Trust, I have passed over the intervening years and the expansion, the successes, and the failures too, that we have experienced in these past 20 years. My endeavour has been to record for you, who do not know the starkness of the contrast that exists between the state of things now and when we started. Progress can never be fast enough for us who have become involved but the record of this organisation is, in my judgement of it, truly worthy of praise.
I leave you then with two cautionary observations. The first is that maybe seeing the massive attendance at this Congress of researchers from all over the world, we may be tempted to the belief that efforts can be relaxed and the research left safely to those in perhaps more affluent countries. One thinks for example of the United States. If so you would be badly wrong. The research contribution made by the United Kingdom Trust is of the very greatest possible importance in the context of international research. As I see it, from my involvement in the Trust, the work done by our researchers, and I include those in England and also Wales, Scotland and Northern Ireland, is regarded as of the very highest quality. Thanks no doubt to those involved but largely I think to painstaking and I would add time consuming care that is given on a voluntary basis by the Members of our Scientific and Medical Advisory Committee to ensure that those projects that are supported by our funds are of sufficient scientific and clinical merit.
If however I may put the matter in terms of not only technical excellence but of hard cash, the following figures may interest you. In the United States, obviously by far the largest member of the international CF community, the total annual sum raised is about 17 million dollars, about 12 million pounds sterling, of which only about one pound out of 12 is spent on research. The reason is that, in the absence of a universal health service such as is available in this country, our US sister body has to devote much of it’s resources to the support of the CF patients and families. You will recognise the importance of the UK research contribution which is the product of our efforts, when you compare that figure of 1 million pounds spent on research in the United States with the figure, not much less, of £700,000 odd spent in the United Kingdom. No doubt some allowance must be made in comparing those figures for the fact that because of higher costs a million pounds buys much less research in the United states than it would in the United Kingdom. So we cannot leave it to others.
My other cautionary observation related to matters nearer home. Under the guidance and, as we all know, exhortation of Ron Tucker the income of the UK Cystic Fibrosis Research Trust has risen progressively over the years. Only once in our 20 years has there been no increase. Usually the sum raised by our groups and branches, which is as you know the major source of income, has increased by 10 to 20% each year. Now Ron Tucker has gone. His successor Charles Spottiswoode has our total confidence and I know he will have our strong support. However, he cannot have instantaneously the same intimate knowledge of our families and of local as well as general CF circumstances. We all know that in the 18 years that he was our Executive Director Ron Tucker developed a close attachment with us all, individual and collectively and with our supporters. It is I believe unavoidable that some of the value of that special relationship will be lost to us with the changeover to a new Executive Director. So I say this. We must all rally to our new Director and give him the extra support that is necessary to sustain our research efforts in this period of transition.
In concluding this history of the Cystic Fibrosis Research Trust therefore, I record my belief that we are potentially in a period where our support could slacken and our income drop. Equally I record my strong conviction that when someone comes to recount the history of the next 20 years it will be found that our membership recognised the possibility a slackening of effort and responded with added vigour. I believe that under the leadership of Charles Spottiswoode we will go on progressing in the way and at the rate we have done in the past and I for one, and I know the members of the Executive Committee have every confidence, that that will be so.
It is my belief too that in our research into improved clinical management and the search for the basic defect spectacular development is imminent After our special history we must not falter now. This Trust, Mr Chairman, has most certainly won its laurels. We who work for it must not rest upon them!
Addendum (Jim Littlewood 2018)
As parents of two children with Cystic Fibrosis, Robert and Linda Johnson were the parents involved in forming the first CF Parents Support Group in London and South East England in 1963, even before the CF Research Trust was officially formed in 1964. In this they were encouraged by Dr Archie Norman whose paediatric clinic their children attended at the Hospital for Sick Children Great Ormond Street in London. Linda and Robert have been continuously involved in one role or another both in the UK and internationally making a major and unrivalled contribution to the care and outlook of people with CF for 50 years. For their contribution to advancing the cause of Cystic Fibrosis both Robert and Linda were awarded Panchaud Medals of the CF Trust – Linda in 1983 and Robert in 2005.
==========================================================
1989 Dodge JA. Cystic fibrosis in Wales. Welsh Paed J 1989; 1:4-6. (www.welshpaediatrics.org.uk/cystic) Full text on the web.
A brief account of paediatric developments in Wales by Professor John Dodge both in terms of clinical care and also mentioning the research carried out there.
1990 Kulczycki LL. Five decades of cystic fibrosis (1938-1988). Acta Universitas Carolinae Medica 1990; 36:7-12.

Fig 19. Lucas Kulczycki celebrating his 100th birthday with Drs. David Nelson and Joseph Bellanti in 2011. ScienceDirect.com
A brief outline of recent history as seen by Dr Lucas Kulczycki (1911-2018) (fig. 19) who worked closely with Harry Shwachman in Boston from 1955 to 1970 until he moved to Georgetown. For many years Lucas Kulczycki was a leading figure in the CF world and published widely on the subject.
“Dr. K,” as he is affectionately known by his colleagues and patients, escaped Poland as the Nazi’s marched through in September 1939 and literally walked over the Carpathian Mountains into Hungary, then to France and eventually across the English Channel to Liverpool and then to Scotland where he received his medical degree from the University of Edinburgh. Dr. Kulczycki then emigrated to Canada to continue his medical studies and then went to Boston where he started to specialize in treating children with a severe respiratory disease known as cystic fibrosis. While in Boston, in 1958, he worked with Dr. Harry Shwachman to devise the Shwachman-Kulczycki Score for determining the severity of a patient’s cystic fibrosis. The Shwachman-Kulczycki Score is a tool still used by medical professionals today. In 1962, Dr. Kulczycki came to Washington, DC where he served as the director of the Cystic Fibrosis Center and a Professor of Pediatrics at Georgetown University School of Medicine. (information and image from the MedStar Georgetown University Hospital website with permission)

Fig 20. Maurice Super
1992 Super M. Milestones in cystic fibrosis. In; Warner JO, editor. Cystic Fibrosis. British Medical Bulletin 1992; 4:717-737.
Maurice Super (fig. 20) wrote a short account of the history of CF which has a strong emphasis towards the genetic advances as he was both a paediatrician and also a geneticist in Manchester, UK.

Fig 21. Margaret Mearns
1993 Mearns MB. Cystic Fibrosis: the First 50 Years. A review of the clinical problems and their management. In: Dodge JA, Brock DJH, Widdicombe JH, editors. Cystic Fibrosis – Current topics. Vol I. Chichester: John Wiley and Sons, 1993: 217-250.
An account of the treatment of cystic fibrosis by Margaret Mearns,(figure 21) a UK paediatrician of great experience and one of the few UK paediatricians who was closely involved with CF for many years from the Fifties, first working with Winifred Young and then as Consultant Paediatrician in charge of the Cystic Fibrosis Clinic at the Queen Elizabeth Hospital for Children, Hackney, London.

Fig 22. Michael Welsh
1998 Welsh MJ, Ramsey BW. Research on cystic fibrosis: a journey from the Heart House. Am J Respir CritCare Med 1998; 157 Suppl: S148-S154.[PubMed]
A review of research in CF by Michael Welsh (fig. 22) , one of the leading US researchers in the field, tracing the “pathway of discovery leading to understanding and cure of a genetic disease”. The stages can be summarised as follows – first the clinical identification of the condition, then description of the physiological/biochemical defect, identification of the gene and mutations, elucidation of the function of the gene product, understanding how mutations cause dysfunction of the gene product, explanation how mutations cause disease and the development of therapies.

Fig 23. Paul Quinton
1999 Quinton PM. Physiological basis of cystic fibrosis: a historical perspective. Physiol Rev 1999; 79:S3-S22 [PubMed] A very clear review by one of the pioneers of CF research particularly putting into perspective the various phases of understanding of the basic defect; in particular. discussing the two apparently distinct faces of CF – that of a mucus abnormality and one with defects in electrolyte transport. Quinton notes that CF “has advanced from innumerable speculations about its cause to a precise definition of causative mutations accompanied by accurate quantitative descriptions of their physiological effects”.
2001 Cystic Fibrosis in the 20th Century. People Events and Progress. Doershuk CF (Ed). A M Publishing Ltd, Cleveland, Ohio. 2001

Fig 24. Cystic Fibrosis in the 20th Century

Fig 24. Carl Doershuk
A very substantial and interesting multi-author account of the developments in CF with a strong emphasis on the North American scene edited by Dr Carl Doershuk 1930-2019) (fig. 24). Carl Doershuk joined Leroy Matthews at the CF clinic in Cleveland in 1960, initially as a Senior Resident, within 3 years of Matthews starting his “comprehensive and prophylactic (preventive) treatment programme” in 1957. There are chapters by many of the those involved in CF in North America, such as Paul di Sant’Agnese, Paul Quinton and many others, looking back with their personal views on the developments in many areas
There is a very interesting review of the book in 2003 by Dr Walter M Robinson, a paediatrician and subsequently an award winning essayist, of the Division of Respiratory Medicine, Children’s Hospital Division of Medcial Ethics,Harvard Medical School, Boston (Pediatric Pulmonology 2003;36:279-282).
Dr Victor Chernick, The Editor in Chief, Paediatric Pulmonology, introduces the review – “Published below is a unique book review that deserves a more prominent site in this Journal rather than being relegated to the back pages and potential obscurity. Cystic Fibrosis in the Twentieth Century is a fascinating story, and the Cleveland Clinic ,Dr Carl Doershuk, and his predecessor Leroy Matthews played a major role in setting the standard care for the CF patient. hence I’ve taken the liberty of putting this review up front.
In the review Dr Robinson congratulates Dr Doershuk and the authors who have done a tremendous service both to current students of CF and to future historians of medicine by providing a detailed source of primary documents in the history of an important childhood disease. That CF has become something that might be altogether unrecognisable to those early clinicians and families is a testimonial to the hard work of scientists and clinicians as well as the perseverance of patients and families . The book should also a source of wisdom and inspiration to those working in the field now and in the foreseeable future.
Dr Doershuk kindly gave me permission to use any illustrations and material from his book – both of which I have used extensively in this cystic fibrosis.online history.
2004 Wellcome Witnesses to Twentieth Century Medicine. Volume 20. Cystic Fibrosis. Christie DA, Tansey EM. (Eds). Wellcome Trust Centre for the History of Medicine at University College. London.

From top down. Prof. John Walker-Smith, Dr Jim Littlewood, Mrs Rosie Barnes and Dr Archie Norman
The transcript of a Witness Seminar held by the Wellcome Trust Centre for the History of Medicine at UCL, London, on 11 June 2002.
The meeting was introduced and co-chaired by Professor John Walker-Smith, a retired Paediatric Gastroenterologist and Dr Jim Littlewood – the then Chairman of the CF Trust’s Research and Medical Advisory Committee.
Some 40 people who had been involved with either the science or clinical aspects of CF spent the afternoon reminiscing on their experiences. In particular, Dr Archie Norman and Sir John Batten both contributed as did Dr Phillip Farrell, who was visiting from the USA at the time.
The editors, Drs Daphne Christie and Tilly Tansey did a magnificent job in producing a valuable detailed account of the many contributions.
2004 The Joseph Levy Memorial Lecture. “Looking back over 40 years and what the future holds”. Littlewood JM.

25a. Gerd Doring, the President, presenting Jim Littlewood with the Rossi Medal in 2004

Fig 25. The 2004 Joseph Levy Memorial Lecture
In 2004 Jim Littlewood was awarded the Joseph Levy Memorial Lectureship by CF Worldwide. Every fourth year the North American and European CF organisations combined and joined with CF Worldwide to put on an international meeting which replaced the annual European meeting. The Joseph Levy Memorial Lecture was delivered at the Opening Ceremony of the 27th European Cystic Fibrosis Conference 2004 in Birmingham, UK. The lecturer usually based his/her lecture on a subject which had been a major interest of their’s over the years. I had by that time had quite a long experience since qualifying in 1956, almost amounting to history! So I choose to review the history of cystic fibrosis much of which I had been involved in and speculate on the future.
A full transcript of the 2004 lecture with references can be downloaded from the UK CF Trust website (www.cysticfibrosis.org.uk) or directly from the Internet (enter Joseph Levy Memorial Lecture 2004)
In 2004, at the same meeting, Jim Littlewood was awarded the Rossi Medal of the European Cystic Fibrosis Society.(fig. 25a).
2008 Fanos JH. “We kept our promises”: An oral history of Harry Shwachman, MD. Am J Med Genet Part A 146A:284-293.[PubMed]

Harry Shwachman.
A really fascinating account of an interview with Harry Shwachman and his wife in 1984 – some 2 years before his death. Harry Shwachman, M.D., was Chief of the Division of Clinical Nutrition at Children’s Hospital, Boston, which eventually became the largest Cystic Fibrosis (CF) center in the world. For over four decades he pioneered understanding of the disease and developed a program of diagnosis and treatment that dramatically extended the lives of children affected with CF. During his own childhood, he developed bilateral pneumonia and nearly died.
Harry Shwachman received a BS from M.I.T in 1932, and an MD from Johns Hopkins in 1936. He completed his internship and residency at Johns Hopkins and Children’s Hospital, Boston (1937–1941). In 1947 he returned from Army service to Boston where he resumed his medical practice and research. Dr. Shwachman discovered a less invasive screening procedure for CF than the intubation technique used previously. In the late 1940s he and his colleagues identified pulmonary involvement to be the primary manifestation of CF. He was instrumental in establishing the autosomal recessive genetic pattern of CF [Allen et al., 1956]. He introduced new treatment methods that changed the management of CF, including the use of antibiotics in 1948, a new pancreatic enzyme replacement, and chest physical therapy. In 1954 Shwachman developed the first reproducible sweat test at Children’s Hospital, Boston, and in 1955 he helped establish the Cystic Fibrosis Foundation. He authored over 250 publications, trained generations of researchers, and was a dedicated clinician, legendary for his devotion to his patients and their families. In addition, he was an accomplished violinist and founding member of the Newton Symphony Orchestra. He and Irene, his wife of 42 years, had three children: Elizabeth, Joan, and Alan, and at the time of interview, three grandchildren. This autobiographical vignette is based on excerpts from an interview I conducted of Dr. Shwachman and his wife in their home in Boston in 1984, 2 years prior to his death. © 2008 Wiley‐Liss, Inc.
2014 Stephanie Clague. Dorothy Hansine Andersen. Lancet Respir Med 2014 Mar;2(3): 184-185. https://pubmed.ncbi.nlm.nih.gov/24621679/

Stephanie Claque
LinkedIn i
A brief interesting account of Dorothy Andersen’s family, professional and private life.
Stephanie Claque is Communications Manager at the Lancet
2016 Wilmott Robert W. An invited article. “A Historical Perspective of Cystic Fibrosis”. Hong Kong Journal Paediatrics 2016;21:280-285.

Fig 25b. Robert W Wilmott
A concise very readable chronological history of cystic fibrosis by Professor Robert Wilmott (fig. 25b). Robert, who qualified in England, is a paediatric pulmonologist in St Louis. After a successful career there he was appointed Vice President for Medical Affairs and Dean of the University School of Medicine, effective from August 2019 until 2021.
Robert Wilmott has served in leadership positions at St Louis University since 2001, including 17 years as Chair of the Department of Pediatrics. He has received numerous awards and has been named a St. Louis “Best Doc” by St. Louis Magazine every year since 2002.
2016 Hodson and Geddes’ Cystic Fibrosis. Fourth Edition Eds. Andrew Bush, Diana Bilton & Margaret Hodson. CRC Press. Taylor and Francis Group.

Hodson and Geddes’ Cystic Fibrosis
The following two chapters in this recent textbook (fig. 26) relate to the history of cystic fibrosis
– Kris De Boeck. Introduction: From the discovery of the gene in 1989 through to 2014 pp 3-17
– James M Littlewood. Appendix A: History of cystic fibrosis. pp 633-649
VARIOUS ARTICLES BY DR RONALD BUSCH
Dr Med. Roland Busch (Medizinisches Zentrum Mitte Rostock, E.-Hilzheimer-Straffe 22-29, Rostock 1, DDR-2500, GDR) has written extensively on the history of cystic fibrosis and reviewed many reports from the Middle Ages onwards of children many of whom may have had the condition. Unfortunately I have not been successful in contacting him although I have reviewed his articles as follows.
1971 Busch R. Zur m der Mukoviscidose beim Neugenboren. (Comprehension of mucoviscidosis in the newborn). Kinderarztl Prax 1971; 39:268-271. (German)[PubMed]
A short article dealing mainly with newborn screening which in 1971 was by examination of the meconium for excess albumin. There is mention of the suggestion of Schutt & Isles (1968, above) to use the Albustix test for detecting the excessive albumin in a solution of meconium.
1978 Busch R. The history of cystic fibrosis. NIH Lib. Trans.53; 316-381.
1978 Busch R: On the history of cystic mucoviscidosis. Deutche Gesundhs.1978; 33:316-320.
This article is in German and contains many references. The English summary: Up to now the casuistry on a newborn that had died of a meconium ileus, its pancreas showing histological typical alterations, published by Karl Landsteiner in 1905 was considered to be the oldest scientific publication. From the literature published before Landsteiner preponderantly cases of gastrointestinal or mixed form of mucoviscidosis may be traced back to the first half of the 19th century. A postmortem record from Vienna in 1838 signed by Rokitansky contains the oldest scientific exactly described presentation in the form of a perforation of the small intestine accompanied by a meconium peritonitis. Besides that by means of an ancient superstition the mucoviscidosis in the folklore area may be traced back till the 17th century.
1979 Busch R. Zur Fruhgeschichte der zystischen Pankreasfibromatose. NTM-Schriftenr Gesch Naturwuiss 1979; 16:95-109. (German).
Only 11 of the 104 references are in English. An extensive review of the previous reports suggesting cystic fibrosis with pictures of Landsteiner, Heubner and Fanconi but interestingly no mention of Dorothy Andersen!
1983 Busch R. Mucoviscidosis (cystic fibrosis), a disease of unclear structure until recently. (German). Gegenbaurs Morphol Jahrb 1983; 129:459-465.[PubMed]
The summary is as follows – “Cystic fibrosis (mucoviscidosis) is described in a review article. The cause of this common lethal hereditary disease of white people is hitherto unknown. The early death of patients with cystic fibrosis, a genetic paradoxon, a lot of hypotheses and very high demands in treatment should challenge more scientists for research. There are added some short notes about the authors work in the history of cystic fibrosis.
1986 Busch R. Historical aspects of cystic fibrosis. Wissenshaftlike zeitsschrift der Willempieck Universistat Rostock. 1986; 35:84-87. 1987 Busch R. Cystic Fibrosis in the XIX century. Arch Hist Filoz Med 1987; 50: 427- 434. (English with Polish summary)[PubMed]
The author investigated the medical literature of the 19th century to detect case records suggestive of the patients having had cystic fibrosis. During the century, not withstanding microscopic techniques, there was little progress in research about the pancreas and meconium. There are useful references to and comments on the German literature dealing with this subject. A number of these reports suggested that the children may have had cystic fibrosis.
1990 Busch R. The history of cystic fibrosis. Acta Univ Carol Med 1990; [PubMed]
Cystic fibrosis is said to have arisen due to a gene mutation 4000 to 5000 years ago (but also see Busch 2005 below). Migration of peoples, gene mutations and new conditions in nourishment could have been the causes. Resulting from old cleaning ceremonies and preventing or treating uncanny effects in children, it was usual to lick the forehead of newborns and children crosswise. If one perceived a salty taste, the child was called bewitched or fascinated and was feared to die soon. The author identified such descriptions in 12 states of modern Europe. Medical documents are reviewed from the first case report until Carl von Rokitansky in 1838 (see below).
1991 Busch R. On the history of cystic fibrosis. Nord Medicinhist Arsb 1991; 95-98.[PubMed]
It is supposed that CF appeared about 3000 BC. Migration of peoples, gene mutations and new conditions in nourishment could have been the cause. Resulting from old cleaning ceremonies and preventing or treating uncanny effects in children, it was been usual to lick the forehead of newborn and children crosswise. If one perceived a salty taste, the child was called bewitched or fascinated and was feared to die soon. The author found describing in 12 states of modern Europe. Medical documents from first case reports are reviewed”.
It is noted that there are no suggestive reports from the United Kingdom and Northern Ireland, nor does the Encyclopedia of Superstitions by Radford and Radford, 1948, contain any key word on cystic fibrosis. The case of Pauw (1595 above) is mentioned also a girl of 3 years treated in 1673 by Georg Seger, a German doctor. Fever, vomiting, diarrhea and wasting for a considerable time before death; the only pathological finding at autopsy was an indurated and scirrhous pancreas
1994 H J Dietzsch. Research and treatment in cystic fibrosis. Eur J Pediatr 1994 Nov; 153(11):783. https://pubmed.ncbi.nlm.nih.gov/7843189/
2005 Busch R. What do we know about the history of cystic fibrosis? Quebec Adult CF Newsletter. 2005; 28-30.
An excellent brief account of many of the earlier references to children who may possibly have had cystic fibrosis. “Around the time when Palaeolithic man left Africa and entered eastern Mediterranean, that is about 52,000 years ago, a gene mutation, the gene mutation responsible for cystic fibrosis first appeared, likely in the Near East” (note also Busch 1990 above re. the dates of the suggested mutation).
2007 Westwood T, Ramsay M. Medical/Scientific topic. Something New Out of Africa: Cystic Fibrosis Affects All Peoples. http://www.cfww.org. CF Worldwide Newsletter Jan 15th 2007.

Fig 27. Michelle Ramsay
An interesting account of CF in South Africa. The article can be downloaded from the website.
Dr. Tony Westwood is a general paediatrician at the Red Cross War Memorial Children’s Hospital in Cape Town. He is the head of the Outpatient and Emergency Departments of the hospital. He is also a senior lecturer in the School of Child and Adolescent Health of the University of Cape Town. He has worked in the hospital’s CF Clinic since 1992 and recently completed a doctoral thesis based on his research on CF in Cape Town and its environs.
Professor Michele Ramsay (fig. 27) is head of the Molecular Genetics Laboratory in the Division of Human Genetics at the

Fig 28. Robert (Bob) Beall
National Health Laboratory Service and the University of the Witwatersrand. Her PhD was followed by a two-year Postdoctoral Fellowship in London, working on the molecular basis of CF. Currently, her laboratory provides a diagnostic service for the molecular investigation of multiple single gene disorders. Her research interests include CF in the black African population, pigmentation in health and disease, hermaphroditism and genetic predisposition to fetal alcohol syndrome.
2009 Robert Steven Kaplan, Sophie Hood. “Bob Beall at the Cystic Fibrosis Foundation”. Published by The Harvard Business School. 9-409-107. April 17th 2009. Copies and permissions from www.hbsp.harvard.edu/education.
This is an interesting account, originally developed for class discussion, of the history and workings of the CF Foundation and its President and Chief Executive Officer Bob Beall (fig. 28) .
2013 Watt P. For the Roses. Cystic Fibrosis Ireland. Published by Cystic Fibrosis Ireland (www.cfireland.ie)

Fig 29. Philip Watt

A very interesting and liberally illustrated account of the history of the Cystic Fibrosis Association of Ireland (now Cystic Fibrosis Ireland) and CF developments there over the 50 years since the organisation was formed in 1963.
The book was written by Philip Watt (fig. 29) the present Chief Executive Officer, who kindly provided with me with a copy. In a letter with the copy Philip writes – “In researching the book and in particular talking to some of those involved in the early days, it quickly struck us that in CF Ireland that we had not done nearly enough to document, acknowledge and thank people who have made such a difference to the CF narrative in Ireland. We were very fortunate that the two women who founded the association were (and remain) alive and well”. Philip refers to Anne O’Dwyer and Bridie Maguire who are widely acknowledged as the two most important people in the establishment and development of the CF Association of Ireland.
2014 Stephanie Clague. Dorothy Hansine Andersen Lancet Respiratory Medicine. 2014 Mar; 2(3):184-5 https://pubmed.ncbi.nlm.nih.gov/24621679
An interesting account of Dorothy Andersen’s Life and family background.
2018 Philip Farrell, Claude Férec, Milan Macek, Thomas Frischer, Sabine Renner, Katherina Riss, David barton, Teresa Repetto, maria Tzetis, Karine Giteau, Morton Duno Melissa Rogers, Hara Levy, Mourad Sahbatou, Yann Fichu, Cedreic Le Marechal , Emmanuelle Genin. Estimating the age of p.(Phe508del) with family studies of geographically distinct European populations and the early spread of cystic European Journal of Human Genetics volume 26, pages 1832–1839 (2018)
Abstract: The high incidence of cystic fibrosis (CF) is due to the frequency of the c.1521_1523delCTT variant in the cystic fibrosis transmembrane conductance regulator (CFTR), but its age and origin are uncertain. This gap limits attempts to shed light on the presumed heterozygote selective advantage that accounts for the variant’s high prevalence among Caucasian Europeans and Europe-derived populations. In addition, explaining the nature of heterozygosity to screened individuals with one c.1521_1523delCTT variant is challenging when families raise questions about these issues. To address this gap, we obtained DNA samples from 190 patients bearing c.1521_1523delCTT and their parents residing in geographically distinct European populations plus a Germany-derived population in the USA. We identified microsatellites spanning CFTR and reconstructed haplotypes at 10 loci to estimate the time/age of the most recent common ancestor (tMRCA) with the Estiageprogram. We found that the age estimates differ between northwestern populations, where the mean tMRCA values vary between 4600 and 4725 years, and the southeastern populations where c.1521_1523delCTT seems to have been introduced only about 1000 years ago. The tMRCA values of Central Europeans were intermediate. Thus, our data resolve a controversy by establishing an early Bronze Age origin of the c.1521_1523delCTT allele and demonstrating its likely spread from northwest to southeast during ancient migrations. Moreover, taking the archeological record into account, our results introduce a novel concept by suggesting that Bell Beaker folk were the probable migrating population responsible for the early dissemination of c.1521_1523delCTT in prehistoric Europe.
2018 Philip Farrell. Discovering the ancient origin of cystic fibrosis, the most common genetic disease in Caucasians. The Conversation. 2018 September 7 (above)
===============================================================
2007 “History of the European Working Group for Cystic Fibrosis & European Cystic Fibrosis Society Conferences” A lecture by Professor Niels Hoiby adapted by Dr Jim Littlewood
This lecture on the history of the European Working Group for Cystic Fibrosis (EWGCF) and the European Cystic Fibrosis Society (ECFS) conferences was given by Professor Niels Hoiby in 2007 at the 30th Annual Conference of the European Cystic Fibrosis Society in Belec, Turkey. We are grateful to Niels for permission to publish the presentation and allow the addition of some additional comment

Fig 1. Ettore Rossi
Professor Ettore Rossi (fig. 1) was Chairman of the Department of Paediatrics of the University of Berne, Switzerland from 1956 until he retired in 1985. He was one of the central figures involved in the development of very many areas of paediatrics in Europe, including cystic fibrosis.
I (Jim Littlewood) met Professor Rossi only briefly on a few occasions – one incident left me with a lasting impression of his kindness and humanity. At an International CF Conference, when he was chairing a plenary session, he had no hesitation in informing one very senior presenter, who had just described a study involving children with CF in numerous annual needle biopsies of the liver, in no uncertain terms, precisely what he thought of the ethics of the study!
Rossi was obviously held in high regard by those who knew him and described as an enthusiastic teacher, a serious hard working paediatrician and a superb medical friend – “the beloved father of hundreds of paediatricians in the whole world”.
In 1969 the first informal meeting of the European Working Group for Cystic Fibrosis (EWGCF) was held at Interlaken, Switzerland and chaired by Professor Rossi. The European Working Party for Cystic Fibrosis was an informal group the founding meeting being held in Cambridge in the UK 1969 with Professor Rossi of Bern, Switzerland (figure 1) as the founding father and first president. The EWGCF only task was to organise annua European CF Conferences in different countries and in that way develop CF care and research throughout Europe. There were no bye-laws. The Secretary, Dr Beat Hadorn is remembered for his early work on pancreatic function tests (Hadorn et al, 1968).

Fig 2. Ron Tucker
The treasurer, Ron Tucker OBE (fig. 2) was the first Director of the UK Cystic Fibrosis Research Trust (now the CF Trust). The second secretary representing Eastern Europe, Vera Vavrova of Prague, was involved in CF care for many years and was awarded the medal of the ECFS in 2008 (figure). Her very interesting presentation “The Story of Cystic Fibrosis in Prague” is available on the ECFS website
Annual meetings of the EWGCF from 1970 to 1977. The President of the conference was usually the senior clinician involved in CF care in that city. (Details from the ECFS website)
1969 Cambridge, UK – Founding Meeting of the European Working Group for Cystic Fibrosis (EWGCF) – E Rossi elected President
1970 1st ECFC – Stockholm, Sweden – Local President/Secretary: Lindquist/Kollberg – Society President/Secretary: E. Rossi/Hadorn
1971 2nd ECFC – London, UK – Local President: Lawson – Society President/Secretary: E. Rossi/Hadorn
1972 3rd ECFC – Reinhardshausen, Germany – Local President: Stephan – Society President/Secretary: E. Rossi/Hadorn
1973 4th ECFC – Warsaw, Poland – Local President: Bozkowa – Society President/Secretary: E. Rossi/Hadorn
1974 5th ECFC – Verona, Italy – Local President/Secretary: Mengoli/Mastella – Society President/Secretary: Mengoli/Hadorn
1975 6th ECFC – Dublin, Ireland – Local President/Secretary: Lyons/Harris – Society President/Secretary: Tempany/Hadorn
1976 No Meeting – 7th International CF Conference – Paris, France
1977 7th ECFC – Dresden, Germany (fig. 7) – Local President/Secretary: Dietzsch/Gottschalk – Society President/Secretary: E. Rossi/Hadorn

Fig 3. Arrowed in the picture at the 1977 Dresden Conference from left to right are Beat Hadorn, Niels Hoiby and Hans -Joachim Dietzsch
By 1977 the EWGCF conferences were becoming larger and there were 170 participants from 19 European countries, USA and Canada, at the Dresden conference of that year.
Professor Hans-Joachim Dietzsch and Dr Bodo Gottschalk worked in Dresden in the German Democratic Republic. In the Eighties Dr Gottschalk visited a number of CF centres in the UK, including our own in Leeds and did not return to Eastern Germany.
EWGCF Conferences 1978 – 1984 During most of this period Manfred Gotz of Vienna (fig. 4) and Richard Kraemer of Berne (fig. 5) were involved as EWGCF Present and Secretary respectively.

Fig 4,. Manfred Gotz

Fig 5. Richard Kraemer
1984 No Meeting – 9th International CF Conference – Brighton, UK
1983 12th ECFC – Athens, Greece – Local President/Secretary: Adam/Manolaki – Society President/Secretary: M. Götz/Kraemer 1982 11th ECFC – Brussels, Belgium – Local President/Secretary: D. Baran/Van Geffel
1981 10th ECFC – Bern, Switzerland – Local President/Secretary: E. Rossi/Kraemer – Society President/Secretary: M. Götz/Kraemer 1980 8th International CF Conference – Toronto, Canada 1979 9th ECFC – Noordwijkerhout, the Netherlands – Local President: Kerrebijn – Society President/Secretary: M. Götz/Kraemer
1978 8th ECFC – Badgastein, Austria – Local President/Secretary: Stur/Götz – Society President/Secretary: Stur/Hadorn

Fig 6. Warren Warwick and Niels Hoiby
At the 1979 Leuwen meeting in the Netherlands, Warren Warwick is seen talking with Niels Hoiby. (fig. 6) Warren Warwick of Minnesota is one of the pioneers of CF care in N. America – in particular, he started the first CF patient registry in the USA in the early Sixties. Warren was still attending CF meetings in 2011. He was also closely involved in the development of the physiotherapy vest eventually used by over half the people with CF in North America.
Warren Warwick was an admirer of the Copenhagen CF centre and wrote in 1982 – “Beginning early in the nineteen-sixties the excellence of the work in pulmonary and cystic fibrosis research done at the Paediatrics Department TG attracted interest from around the world and cystic fibrosis researchers from everywhere began to visit Denmark to learn from Doctor Erhard Winge Flensborg. I joined that group of visitors in 1966 when I wrote to Doctor Flensborg for permission to visit his department. Like all the others, I was welcomed. The intellectual stimulation there surrounded by the sparkling hospitality of Erhard and Inga Flensborg and the doctors studying at Paediatric Department TG was so helpful that I have eagerly returned on many occasions over these past 16 years, as have others who have made a first visit to Paediatric Department TG”.
Niels Hoiby (fig. 7) had entered the CF field in the early Seventies in Copenhagen, where a CF centre had been started in the mid-Sixties by Erhard Winge Flensborg. By 2023 Niels was the author or co-author of over 500 publications, most on the microbiological and immunological aspects of cystic fibrosis.
EWGCF Conferences 1985 – 1990 From 1987 Niels Hoiby (fig. 7) was the President and Martin Schoni (fig. 8) the secretary of the EWGCF.

Fig 8. Martin Schoni

Fig 7. Niels Hoiby
1985 13th ECFC – Jerusalem, Israel – Local President/Secretary: Katznelson/Godfrey – Society President/Secretary: M. Götz/Kraemer 1986 14th ECFC – Budapest, Hungary – Local President/Secretary: Boda/Gyurkovits – Society President/Secretary: M. Götz/Kraemer 1987 15th ECFC – Oslo, Norway Local President: Michalsen – Society President/Secretary: N. Høiby/M. Schöni 1988 Meeting – 10th International CF Conference – Sydney, Australia
1989 16th ECFC – Prague, Czech Republic – Local President/Secretary: Houstek/Vávrová – Society President/Secretary: N. Høiby/M. Schöni
1990 Joint meeting with the North American CF Conference – Washington, USA
1986 the 1st Latin American CF Congress in Buenos Aires, Argentina. (fig. 9)

Fig 9. Omar Pivetta (Arg) Robert D McCreery(USA), Niels Hoiby (DK), John Mangos (USA) 1986 Buenos Aires
Omar Pivetta is a local paediatrician. Robert (“Bob”) McCreery was President of the International Cystic Fibrosis (Mucoviscidosis) Association (ICF(M)A). (fig. 9). He had been elected in 1976 and for many years was a major international figure on the CF scen
John Mangos (fig. 9) from the USA was a leading CF researcher and paediatrician – he was also a priest. It is reported that he was once asked to see a patient while still in his priest’s robes. The startled patient awoke and declared “Surely I’m not that bad!!”
Robert and Linda (fig. 10) (later Sir Robert and Lady Linda) Johnson had two children with CF and were involved with the UK CF Research Trust from the early Sixties and later with the ICF(M)A. Sir Robert only retired as Vice Chairman of the UK CF Trust in 2009. Both were awarded the John Panchaud medal of the CF Trust for their work.

Fig 10. Linda Johnson (UK),George Adams (GR), Paul Quinton (USA), Inge Saxon Mills (Italy)
George Adam (sitting in the fig. 10) was a leading paediatrician in Athens and health issues among children called involved in CF care in Greece. He was President of the 1983 EWGCF conference in Athens. He had followed Dr Maria Nicolaidou who had been a pioneer of CF care in Greece, after she returned to Athens in the Sixties having visited Dr Harry Shwachman in Boston.
Paul Quinton (standing at the desk in fig. 10) is a leading US scientific researcher, who described the impermeability of the sweat gland duct to chloride, regarded as one of the key scientific discoveries of the Eighties. Paul Quinton has cystic fibrosis.
THE CYSTIC FIBROSIS GENE WAS IDENTIFIED IN 1989

Fig 11. Presentation of the Paul di Sant’Agnese Award to the leaders of the teams who identified the gene – fro L. to R. are Lap Chi Tsui, Paul di Sant’Agnese, Evelyn Graub, Milton Graub, Francis Collins and Jack Riordan
There was a memorable NACFC Conference held in the autumn of 1989 soon after the publication of the three historic papers in Science reporting the identification of the CF gene (for more details see “1989 Science”). There was a general air of euphoria that it would only be a short time before gene therapy would be available for patients – unfortunately within a few years the difficulties soon became apparent and even by 2011 gene therapy was still not available for the patients. However, the UK Gene Therapy Consortium of clinicians and scientists in London, Oxford and Edinburgh embarked on a multi dose year-long trial of gene therapy in 2012.This showed some effect but not sufficient to recommend as a treatment. However, by 2018 the UKGTC were preparing to embark of a further gene therapy trial this time using a more efficient viral derived vector. However, by 2023 there was still no gene for cystic fibrosis.
In 1965 the International Cystic Fibrosis (Mucoviscidosis) Association (ICFMA), the predecessor of CF Worldwide, was formed in Paris under the medical chairmanship of Paul di Sant’ Agnese.
At this time the EWGCF was contributing to the work on ICF(M)A grants. In 1960, prior to his moving from New York to The National Institutes of Health, di Sant’Agnese visited Europe to meet other clinicians interested in cystic fibrosis. These included Prof. Guido Fanconi and Prof. Ettore Rossi (Switzerland), Prof. Andre Hennequet and Dr Jean Feigelson (France), Drs Archie Norman and David Lawson (England), Prof. G de Toni (Italy) and Profs. Wejiers, Dicke and Van de Kamer (Holland).
The aims of the ICF(M)A were to improve the care of children and adults who had CF, to foster research and to disseminate information. Also di Sant’Agnese notes that prior to this meeting those working in CF did not know each other except through the literature and so the ICFMA brought together physicians, paramedical personnel and lay people giving them a sense of common purpose. This was the start of the International CF (ICFMA) now CF Worldwide meetings, which occur every 4 years. It is difficult to appreciate the problems with communication and travel in the Sixties – before the days of easy air travel, photocopiers, fax machines, e-mail, internet and medical databases such as Medline and Pubmed. So the formation of the ICF(M)A was undoubtedly a major milestone.

Fig 12. ICF(M)A course in Petersburg, 1992. Christian Koch, Niels Hoiby, A Hansen, Russian Dr T Guembitskaya, V Baranov
On the left of this group in St Petersburg (fig. 12) are Christian Koch (paediatrician) and Niels Hoiby (microbiologist), respectively the clinical and scientific leaders of the very successful Danish CF Centre in Copenhagen.
In the previous year a seminal publication from their centre (1991 Valerius NH, Koch C, Hoiby N. Prevention of chronic Pseudomonas aeruginosa infection in cystic fibrosis by early treatment. Lancet 1991; 338:725-726) had reported that early Pseudomonas aeruginosa infection could be eradicated by a 3 week course of inhaled colomycin and oral ciprofloxacin. Undoubtedly one of the more important papers in the CF literature up to that ti
1988 10th International CF Conference Sydney Australia EWGCF Conferences 1991-1997
EWGCF Conferences 1991-1997
1991 Copenhagen, Denmark Hoiby/Schoni (Hoiby/Pedersen) 1992 International Meeting Dublin Eire 1993 Madrid, Spain Hoiby/Schoni (Escobar/Baquero/Suarez)
1994 Paris, France Hoiby/Schoni(Navarro)
1995 Brussels, Belgium Hoiby/Schoni (Baran/Dab)
1996 International Meeting – Jerusalem
1997 Davos, Switzerland EWGCF Hoiby/Schoni (Schoni/Kraemer/Rossi) ECFS Hoiby/Ryley

Fig 13. Jean Fiegelson, Paris. N Kapranov, Moscow. Martin Weibel Switzerland, President of ICF(M)A. EWGCF Copenhagen 1991
Jean Feigelson of Paris (1921 – 2017) (fig. 13 upper) is one of the pioneers of CF care and still, in 2010 attending CF conferences in Europe and North America. He trained in paediatrics at the Sick Children’s Clinic in Oslo in 1952. In a career spanning over 45 years he has treated over 250 people with cystic fibrosis.
Jean Feigelson’s first recorded paper on cystic fibrosis was in 1960 (Feigelson J. The treatment of mucoviscidosis or cystic fibrosis of the pancreas. Bull Mem Soc Med Paris 1960; 9:4-17); his most recent is as a co-author of a paper on partial splenectomy, which was published in 2007 (Louis D et al, Pediatr Pulmonol 2007; 42:1173-1180). He has 48 references noted in Medline produced steadily over 40 years
Dr Kapranov (fig. 13) presented an account of CF developments in Russia at a meeting of the Royal Society of Medicine in 1995. (Cystic Fibrosis in Russia: background and a model for future collaboration with the West. Kapranov N, Ginter E, Kashirskaja N, Hill CM, Ilangovan P, Rolles CJ. J R Soc Med. 1996;89 Suppl 27:44-7. Review. No abstract available). The first case of CF was diagnosed in Russia in 1967 and it was not until 1989 that the first two CF centres opened, the first in Moscow and the second in St Petersburg.
A more recent account (2007) on the “Management of Cystic Fibrosis in Russia” by Kashirskaya N.Y and Kapranov N.I. from the Russian CF Centre, Research Centre for Medical Genetics, RAMS, Moscow, Russia, is published on the internet under that title.

Fig 14. EWGCF Bruxelles 1993. David Baran, Niels Hoiby, Isi Dab
Professor Isi Dab (fig. 14) made many important contributions to CF (Malfroot A, Dab I. New insights on gastro-oesophageal reflux in cystic fibrosis by longitudinal follow up. Arch Dis Child 1991; 66:1339-1345). Gastro oesophageal (GO) reflux was first described as a problem in CF by Jean Feigelson (Feigelson et al, 1975) and subsequently the problem was thought to be related to progressive CF in older children. However, in the Belgian study from Anne Malfroot and Isi Dab, 21 of 26 (81%) young children with CF aged less than 60 months were studied and 20 confirmed to have reflux by oesophageal pH tracings. Sixteen improved with anti-reflux treatment with improved weight gain, less cough and wheeze but half still had the reflux one year later. The authors concluded the reflux was not caused by the CF chest problems as it improved with time – at the same time as the CF gets worse – hence their title “new insights into GO reflux”.
Professor David Baran (fig. 14) from Brussels has published on a wide variety of topics related to CF since 1970 most recently on the use of N-acetylcysteine (App EM, et al. Eur Respir J. 2002;19:294-302).

Fig 15. ECFS Paris 1994. Henry Riley

Fig 16. EWGCF BRUXELLES 1995. C Moser, H Krogh Johansen (DK)
Dr Henry Riley,(fig.15) the microbiologist from Cardiff, has been involved in CF care and research for many years. He has also been involved in various senior roles with the EWGCF and then the ECFS. In 2023 Henry is still is active in the organising current references for the production of the ECFS newsletter.
1997 THE EUROPEAN CYSTIC FIBROSIS SOCIETY WAS FOUNDED IN DAVOS
 European CF Society Conferences 1998 – 2005
European CF Society Conferences 1998 – 2005
1998 Berlin, Germany Hoiby – Gerd Döring (WahDöring) elected new President
(Van der Laag/Heijerman)
1999 Latin American CF Congress Buenos Aires
2000 International Stockholm, Sweden
2001 Vienna, Austria Döring(Gotz) 2002 Genoa, Italy Döring (Romano)
2003 Belfast, N Ireland Döring (Elborn)
2004 Birmingham UK Döring (Littlewood/Geddes)
2005 Crete, Greece Döring (Nousia-Arvanitakis)
Marie Johannessonn was elected President starting after the 2006 congress
1999 First Consensus Conference, Artimino, Florence

Fig 18. First Artimino Conference in 1999

Fig 18a The Artimino Nutritional Conference
Gerd Döring arranged a number of consensus conferences in Artimino, Italy (fig. 18) A subject was chosen and preliminary documents prepared by leading workers in the particular area. A meeting was then convened of about 30 participants who had a strong interest and expertise in the topic under discussion and a consensus statement was developed at the meeting over 2 days. A report was then prepared and submitted for publication. Most of these consensus documents are available on the ECFS website as free downloads.
These conference that Gerd arranged proved to be a great success both from the consensus documents that resulted but also they were a very pleasant social occasion where delegates and the relatives could get together in a very pleasant informal environment.
European Cystic Fibrosis Society Conferences 2006-2012
2006 Copenhagen, Denmark Döring (Hoiby)
2007 Belek, Turkey Johannesson(Gocmen)
Marie Johannesson retired on the grounds of ill health and was succeeded by Stuart Elborn as President
2008 Prague, Czech Republic Elborn (Pohunek/Macek)
2009 Brest, France Elborn (Ferec/Lehn)
2010 Valencia, Spain Elborn (Sole/Vasquez)
2011 Hamburg, Germany Elborn (Ballman/Mall)
2012 Dublin Elborn (McElaveny/McKone/Plant
2013 Lisbon, Portugal Elborn (Amaral/Barreto)
2004 “New Frontiers in Basic Science of Cystic Fibrosis”

Fig 19. Margarida Amaral
In 2004, the ECFS began another annual conference which is entirely dedicated to basic science. The “New Frontiers in Basic Science of Cystic Fibrosis” Conferences are characterised by active discussion of data and ideas at the forefront of research on CF and CFTR, in an informal, co-operative environment. The ECFS acknowledge the enormous contribution of Prof. Margarida Amaral made in establishing these (fig. 19)
Two stalwarts of the European Working Group for Cystic Fibrosis and later the European Cystic Fibrosis Society receiving the well-deserved ECFS Awards from the President, Stuart Elborn.
The 2011 ECFS Award was presented to Prof. Gerd Döring (Germany) (fig. 21) in recognition of his outstanding contribution to unravelling of the complexities of infection and innate immunity in the CF lung and translating this knowledge into better treatment for people with CF. The ECFS also wished to recognise and honour the huge contribution he has made to the development of scientific research and clinical care in Europe through his leadership of the European Cystic Fibrosis Society.
The 2012 ECFS Award was presented to Professor Niels Høiby (Denmark) (fig. 20) in recognition of his outstanding contribution to the understanding of Pseudomonas lung infection and translating this knowledge into better outcomes for people with cystic fibrosis. He was also recognised for his contribution as the Founding President of the European Cystic Fibrosis Society and his lifelong commitment to the CF center in Copenhagen.
Subsequent ECFS Awards
-

- Fig.21 Gerd Döring 2011
-

- Fig.20 Niels Hoiby 2012
-

- Fig.23 Eitan Kerem 2013
-

- Fig 22. Kevin Webb 2013
-

- Fig 24 Carlo Castellani 2015
-

- Fig 25. Di Bilton 2016
-

- Fig 26 jeffrey Beekman 2017
-

- Fig 27 Sue madge 2018
-

- Fig 28 Stuart Elborn
-

- Fig. 29 Kris de Boeck