Mega papers
Some of the publications that have had a major influence on the understanding and/or treatment of cystic fibrosis. We welcome comments and suggestions for additional “megapapers”..
1929 was the start of the antibiotic era that would lead to the development of penicillin in the Forties. Few people with CF survived early childhood before antibiotics became available.
1929 Flemming A. “On the antibacterial action of cultures of a penicillium, with special reference to their use in the isolation of B. influenza.” Br J Exp Pathol 1929; 10: 226–36.

Fig 1. Sir Alexander Flemming
Alexander Flemming (1881-1955) (figure 1) was a Scottish bacteriologist, working at St Mary’s Hospital, London, whose discovery of penicillin (1928) prepared the initial step towards the highly effective practice of antibiotic therapy for infectious diseases.
In 1945 Fleming shared the Nobel Prize for Physiology or Medicine with Ernst Boris Chain (1906-1979) and Howard Walter Florey (1898-1968) who both (from 1939) were responsible for carrying forward Fleming’s initial observation by further isolation, purification, testing, and quantity production of penicillin
In 1940 a report was issued describing how penicillin had been found to be a chemotherapeutic agent capable of killing sensitive germs in the living body. Thereafter great efforts were made, with government assistance, to enable sufficient quantities of the drug to be made for use in World War II to treat servicemen with war wounds.
Penicillin became available for a few patients with CF in the USA in 1943 and the results were reported by Paul di Sant’Agnese (1944 below) – prior to this virtually all children with CF died in infancy or early childhood from Staphylococcal pneumonia and malnutrition.
It is noteworthy that Flemming failed to develop his 1929 discovery – this was done over 10 years later by Florey and Chain. Nor was Flemming the first to observe the antibacterial effects of moulds.

Fig 2. Ernest Duchesne
Ernest Duchesne (1874 –1912) (figure 2) was a French physician who noted that certain moulds kill bacteria. He made this discovery thirty-two years before Alexander Fleming observed the antibiotic properties of penicillin, a substance derived from those moulds, but his research went unnoticed. Duchesne entered l’Ecole du Service de Santé Militaire de Lyon (the Military Health Service School of Lyon) in 1894. Duchesne’s thesis, “Contribution à l’étude de la concurrence vitale chez les micro-organismes: antagonisme entre les moisissures et les microbes” (Contribution to the study of vital competition in micro-organisms: antagonism between moulds and microbes), that he submitted in 1897 to get his doctorate degree, was the first study to consider the therapeutic capabilities of moulds resulting from their anti-microbial activity. Duchesne had made his breakthrough by observing how the Arab stable boys at the army hospital kept their saddles in a dark and damp room to encourage mould to grow on them. When he asked why, they told him that the mould helped to heal the saddle sores on the horses. Intrigued, Duchesne prepared a solution of the mould
In a series of meticulous experiments, Duchesne studied the interaction between Escherichia coli and Penicillium glaucum, showing that the latter was able to completely eliminate the former in a culture containing only these two organisms. He also showed that an animal inoculated with a normally lethal dose of typhoid bacilli would be free of the disease if the animal was also inoculated with Penicillium glaucum. Unfortunately, as he was only 23 years old and unknown, the Institut Pasteur did not even acknowledge receipt of his dissertation! He urged more research but unfortunately his army service, after getting his degree, prevented him from doing any further work. Considerable attention has been allotted to penicillin as before the availability of penicillin few infants with CF survived infancy. It was the introduction of penicillin and later other antibiotics which was the main development that permitted survival beyond infancy.
1922 Discovery of insulin would eventually be important in the control of CF related diabetes mellitus that eventually would affect the majority of adults.
1922 Banting FG, Best CH. Internal secretion of the pancreas. J Lab Clin Med 1922; VII: 251-266.

Fig 3. Sir Frederick Banting
The work reported in this classic paper eventually led to a Nobel Prize in 1923 for Banting and Macleod, in whose Toronto laboratory the work was done. Frederick Banting (1891-1941) (figure 3) was a Canadian orthopaedic surgeon who became an assistant in physiology in Ontario where he worked in Richard McLeod’s laboratory with Charles Best, a medical student. It was here, after many ups and downs and arguments and with the help of Bertram Collip, a highly trained biochemist (to extract the insulin) that they eventually produced and tried the insulin on a diabetic patient in 1922.
The paper begins –“The hypothesis underlying this series of experiments was first formulated by one of us in November 1920 (Banting was then assistant in Physiology at Western University, London, Ontario) while reading an article dealing with the relation of the isles of Langerhans to diabetes (Barron M: The relation of the islets of Langerhans to diabetes with special reference to cases of pancreatic lithiasis. Surg Gynec Obstetr 1920; xxxi :437-448). From the passage in the article which gives a resume of degenerative changes in the acini of the pancreas following ligation of the ducts, the idea presented itself that since the acinous but not the island tissue degenerates after this operation, advantage might be taken of this fact to prepare an active extract of the islet tissue. The subsidiary hypothesis was that trypsinogen or its derivatives was antagonistic to the internal secretion of the gland. The failures of other investigators in this much worked field were thus accounted for”.
The authors concluded from their experiments that –“intravenous injections of extract from dog’s pancreas, removed from 7 to 10 weeks after ligation of the ducts, invariably exercises a reducing influence upon the percentage sugar of the blood and the amount of sugar excreted in the urine” This medical classic is a “good read” and one of the most significant medical papers of the 20th century as also is Moses Barron’s paper that first gave Banting the idea (above).
The present paper is included here not only for its historical interest but also for its eventual relevance to CF as the majority of those affected will eventually develop CF related diabetes in later life. Although the Islets of Langerhans in CF are functioning adequately through childhood in most patients, despite their severe exocrine pancreatic insufficiency, they are eventually destroyed resulting in the majority of adults with CF developing diabetes mellitus.
Efforts to prevent the slow destruction of the pancreas and prevent or delay the onset of CF related diabetes will surely become an area of research as more people with CF survive to develop this complication which has an adverse effect both on their prognosis and quality of life. It is of relevance and encouraging that treatment of people with CF and a G551D mutation with ivacaftor has been reported to improve insulin secretion (Bellin et al, 2013 below).
1936 Fanconi’s paper is included only as it is considered by many Europeans to be the first clear description of cystic fibrosis although it certainly did not lead to widespread recognition of the condition
I am grateful to my friend and erstwhile St James’s surgical colleague Mr Archie Crompton for translating this article for me.

Fig 4. Guido Fanconi
Guido Fanconi (1892-1979) (figure 4) was Professor of Paediatrics in Zurich from 1929-1965 and a pioneer of modern scientific paediatrics. Although this publication is considered by some Europeans to be the first clear description of CF, it is a brief report of only two children who died aged 10 months and three years with ‘coeliac syndrome’, purulent bronchitis and bronchiectasis. The pancreatic changes were quite typical of those later described in cystic fibrosis. “The changes in the lungs and pancreas, two vital organs, are so profound that their failure appears understandable”.
Martin Bodian quotes a 1928 publication of Fanconi’s (1928 Beih. Jb. Kinderhlk 21) as noting cases of coeliac disease starting in early infancy and often associated with bronchitis but surprisingly Fanconi did not mention this in this 1936 paper which is so frequently quoted in Europe as the first description of cystic fibrosis.
Speaking in 2008, Walter Hitzig, the distinguished immunologist from Zurich, recalls (referring to this paper) that “My teacher Guido Fanconi, in his usual enthusiasm, presented a “new disease” to the Swiss paediatricians in 1936. However, one of the doctors questioned its importance in his daily practice. Fanconi’s answer: “Oh certainly it is important – I have seen already 3 cases!” of course earned him a big laughter in the audience! And today the syndrome described as Pankreas-Fibrose, now called cystic fibrosis, is considered to be the most frequent hereditary disease in the Caucasian population, and no doubt every paediatrician must diagnose it”.
Lynn Taussig describes meeting Knauer, the third author of this paper, after it was published. Apparently the two children described in Knauer’s doctoral thesis in 1935 were the two described in this paper and the next year Fanconi, Uehlinger and Knauer published this paper. He notes that the first name on the paper was now Fanconi’s!
Maurice Super mentions that there was controversy between Fanconi and Wangensteen “who thought the condition might be something unique to the cantons in which Fanconi worked”. The hereditary nature of CF was not suspected at this time.
Fanconi published a number of papers on various aspects of intestinal malabsorption and other aspects of paediatrics between 1921 and 1956 mostly in German or French. He was referred to in one article as a “Jack of all trades” – as were many of the paediatricians of the time. Reviewed objectively, none of these publications, with the possible exception of this 1936 paper, made any major or original contribution to the understanding or definition of cystic fibrosis. However, Fanconi made other very important contributions; in 1929 he described hereditary panmyelopathy known as Fanconi’s anaemia, in 1941 he deduced that polio was spread via the gastrointestinal route rather than by droplets and apparently predicted that Down’s syndrome was due to a chromosome abnormality 20 years before trisomy 21 was discovered
Addendum 2020. Jurg Barben. First description of cystic fibrosis. J Cyst Fibros 2021; 20(1): 183

Fig 4a Jurg Barben
Professor Jurg Barben (Fig.4a) discussing the widespread cited article by Dorothy Andersen in 1938 as the first description of cystic fibrosis, draws attention to the fact that Guido Fanconi’s described two children in 1936 who almost certainly had cystic fibrosis as judged by the autopsy findings. In addition to Andersen’s substantial numbers, her article was in English and Fanconi’s was written in German and published in a German language journal (The Vienna Medical Weekly). Barben observes this publication has now been forgotten in the English-speaking world. However, a German-American colleague, Dr Teodore Bruns (former CF Centre Director in Milwaukee, USA) did a personal translation of Fanconi’s publication many years ago which was never published. This English translation in now (with Permission of Phillip Farrell, Emeritus Dean and Professor for Pediatrics and Population Health Services at the University of Wisconsin, USA who provided the translation) available on line in order to preserve it for the next generation of researchers. https://www.kispisg.ch/downloads/kompetenzen/pneumologie/fanconi-1936_english-translation.pdf
1938 Second of three papers by Margaret Harper of Sydney who clearly recognised pancreatic steatorrhoea – but not clearly enough in her earlier 1930 paper to cause others to recognise CF as a specific condition
1938 Harper MH. Congenital steatorrhoea due to pancreatic defect. Arch Dis Child 1938; 13:45-56.

Fig 5. Margaret Harper
Further to her 1930 report of two children with steatorrhoea (Harper, 1930 above), Margaret Harper of Sydney reported a further eight children with features compatible with cystic fibrosis. Congenital steatorrhoea, abnormal glucose tolerance curves and characteristic pancreatic lesions in all the 4 who were autopsied; 8 of the 10 who died had bronchopneumonia. “The passage of fatty stools from birth (particularly “butter stools”) with failure of nutrition, the diagnosis of congenital defect of the pancreas can reasonably be made”. Subsequently Harper reported 42 children with pancreatic steatorrhoea from the Royal Alexandra Hospital for Children, Sydney (Harper,1949 below). Interesting comments on Andersen’s paper in this 1949 paper (below).
The orange-yellow oil in, on and around the stools is now recognised as a typical and frequent feature of untreated cystic fibrosis. The discussion in this paper reviews previous reported cases with particular emphasis on the characteristics of the stools. A visit to the ward sluice to inspect the stools of children on the ward was a usual part of the paediatric ward round even in the Sixties and considered to be an essential part of the full evaluation of an ill child just as was chemical testing and microscopy of the urine by the ward doctor.
I am grateful to Professor John Walker-Smith for further information on Margaret Harper (fig. 5). He notes that she was a pioneer of women in medicine and reported to have been the first lecturer in Diseases of the Newborn to be appointed in the British Empire. She was appointed as a full time paediatrician in 1914 at the Royal Alexandra Hospital for Children in Sydney thus may be one of the first full time paediatricians.
Writing in 1979, Dr D G Hamilton described Margaret Harper (fig. 5) as – “the brilliant outstanding mind at the Royal Alexandra Hospital for Children. She continued her interest in babies and their welfare throughout her whole life. She greatly influenced the care of children with gastroenteritis with feeding problems and with chronic diarrhoea. These latter were grouped under the title of the Coeliac Syndrome. Last century Samuel Gee in Britain had talked of offensive bread when discussing these children. Margaret Harper by patient trial of diets always seemed to achieve better results with her coeliac patients than any other physician and one of the things she did was to avoid giving them cereals. They were fed copious amounts of banana and cottage cheese. In 1945 it was recognised in Holland that one of the more common causes of chronic diarrhoea in little children is intolerance to gluten that is found in wheat, rye, barley and oats and the name coeliac disease was given to this particular condition. Margaret Harper’s diet of bananas, cottage cheese and no cereal was close to the mark. She was a very careful and thoughtful observer. Among these babies she recognised that a small group were different from the others. They were more prone to chest infections and their stools looked to her to be different and more greasy. These children were prone to die. She drew the attention of Dr Tidswell the director of pathology, to her belief that they were different from the others and at post-mortem he found characteristic changes in the pancreas. She was the first person in the world to recognise this as a separate condition and in 1930 published a report in the Medical Journal of Australia entitled “Two cases of congential pancreatic steatorrhoea with infantilism”. Similar reports followed in 1935 by Arthur Parmalee in Chicago. In 1938 Margaret Harper published a more detailed paper on the subject in the British Archives of Diseases of Childhood entitled Congenital steatorrhoea due to Pancreatic defect.
In 1954 Dr Charles May, an American expert, wrote a monograph on Cystic Fibrosis of the Pancreas (see May 1954 below) and dedicated the work to “the practitioners Margaret Harper of Sydney, Australia and Arthur H Parmalee of Chicago, Illinois who recognised…..and published the first papers, indicating the frequency and importance of the disease, clearly setting it apart from Coeliac disease, against prevelance practice.
Apparently Margaret Harper was always a much loved figure at the hospital, tall, very helpful to those who tried hard, always prepared to be critical, but tempering her criticism with a laugh. She had a splendid scientific mind. She entered happily and freely into bedside discussions and informal argument at clinical meetings where she was usually extremely stimulating, but she hated formal lectures. These she simply read from a prepared script in a monotonous tone and with her head down. She really was a dreadful lecturer. But she was perhaps the greatest physician the hospital has known” (DG Hamilton. Hand in Hand. The Story of the Royal Alexandra Hospital for Children, Sydney. John Ferguson, Sydney. 134-136:1979)
1938 The paper by Dorothy Andersen undoubtedly was of a size and contained sufficient detail to allow others to recognise cystic fibrosis as a definite clinical entity as evidenced by the many subsequent reports that soon followed
1938 Andersen DH, Cystic fibrosis of the pancreas and its relation to celiac disease: a clinical and pathological study. Am J Dis Child 1938; 56:344-399.
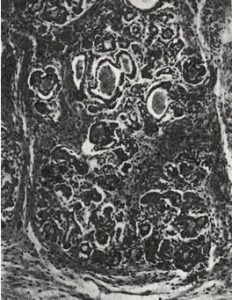
Fig 7. Pancreas CF infant at 6 day
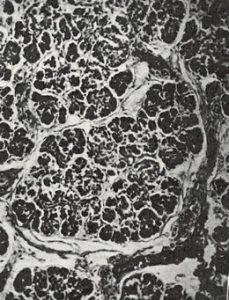
Fig 6. Pancreas non-CF infant at 3 days
Although many infants had already been reported who, in hindsight, obviously had cystic fibrosis, this was the first clear detailed clinical and pathological description of a large series of affected infants in English. Dorothy Andersen reported 49 patients – 20 from her hospital and others from colleagues and the literature. She described the neonatal intestinal obstruction, intestinal and respiratory complications and many other features – but particularly the characteristic pancreatic histology (figures 7 & 8).
Salient points from the original description are as follows – “In 45 of the cases the pancreas presented a microscopic picture which is described by the term cystic fibrosis. The acini contain secretions of various sizes, and the acinar cells were flattened to form a thin epithelial wall around them. The smaller concretions were surrounded by relatively normal cells, which occasionally contained eosinophil granules…….The size of the cysts varied in each case but large ones were not often noted in the youngest infants. Surrounding the acini and also the lobules there were moderate to large amounts of fibrous tissue, the quantity varying roughly with the age of the child…….The islets of Langerhans were usually normal in number and appearance.”
Andersen likened much of the epithelial histology to that found in vitamin A deficiency which for some years she regarded as the primary cause of the systemic features of the condition; this causation was never substantiated although she continued to support this theory for some years.
The paper contains an excellent tabulated review of the previously reported cases and describing her paper Martin Bodian (1952 below), the Great Ormond Street pathologist, writes – “such a clear account of the symptoms that it enabled many cases to be recognised that had hitherto been missed, and aroused such interest that it was followed by a shoal of case reports and confirmatory reviews” which appeared during the Forties” (many are described below).

Fig 8. Dorothy Andersen
Dorothy Andersen (1901-1963) (figure 8) was the pathologist at the Babies Hospital at the Columbia Presbyterian Medical Center in New York. She had a wide range of interests and made contributions in many other areas of medicine. She was said to be a rugged individualist, a paediatric clinician, pathologist, a research chemist – also apparently a roofer and carpenter, happy to make her own home improvements! Eventually over 600 children with CF were referred to her in New York; she involved Dr. Paul di Sant’Agnese, eventually another leader in the CF field, to provide paediatric care for her patients as she was primarily a pathologist.
Dr Phillip Farrell of Wisconsin lists 4 reasons for the referral of so many children to her in New York. First was this seminal 1938 publication, second a combination of her reputation and her arrogance (she is alleged to have said she was the only person in the world who knew about cystic fibrosis in the late Thirties and Forties!), third the desperation of parents and fourth New York was the one place people could reach easily in those days.
Described as “windblown” by friends and detractors alike, Dorothy Andersen was considered quite a character. She is said to have kept a particularly untidy lab, holding semi-annual “glüg” parties there, in honour of her Scandinavian heritage. She was a niece of Hans Christian Andersen. She was described as a bright compassionate and sensitive scientist who was also a chain smoker and died of lung cancer at the age of 62 years.
In 1950 Milton Graub, a paediatrician who eventually became President of the US CF Foundation, suspected his two year old son had CF and describes how he called Dr Andersen on a Sunday morning and found her working in her laboratory. She listened to the medical history and saw his son the next day. The diagnosis was much more difficult and was made primarily on the clinical features plus analysis of the duodenal secretions. These were done and the diagnosis of cystic fibrosis of the pancreas was established. Since Dr Andersen was a pathologist she referred his son Lee for further clinical care to Dr Paul di Sant’Agnese. “He was a pediatric specialist and a delight – soft spoken, reassuring and supportive” (from Doershuk CF, 2001. below).
Finally, her name is frequently misspelt as “Anderson”even by distinguished writers!!
1938 As in the case of Dorothy Andersen’s 1938 paper, recognition of similar characteristic changes in the pancreas of occaisional infants at autopsy suggested a specific condition
1938 Blackfan KD, May CD. Inspissation of secretion and dilatation of ducts and acini, atrophy and fibrosis of the pancreas in infants. A clinical note. J Pediatr 1938; 13:627-634.
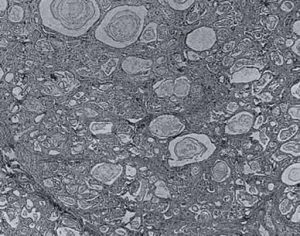
Fig 9. Pancreas with inspissated secretion, dieted ducts and atrophy. Blackfan & Wolbach 1933.
Over a period of 15 years, examination of the pancreas in over 2,800 paediatric necropsies in Boston disclosed 35 examples of a well-defined pathologic lesion the existence of which was unsuspected during life. The lesion was characterised by inspissation of secretion, dilatation of the ducts and acini, atrophy and fibrosis. Only seven showed the stratified epithelium characteristic of vitamin A deficiency. The need for further information concerning the absorption of fat and especially nitrogen as well as pancreatic enzymes as an aid to diagnosis was suggested. The average age of death was 8 months.
Blackfan & Wolbach (1933 above), from a study of the lesion in the large amount of material available, in regard to pathogenesis, had observed “Our preliminary studies indicate that the pathogenesis of this striking pancreatic affection (figure 9) resides in the production of an abnormal secretion which inspissates and leads to distension and atrophy of the ducts and acini” (Blackfan & Wolbach, 1933; May CD 1943).
Dr. Charles D. May (1908 – 1992) was a paediatrician, an expert on childhood nutritional disorders and a leading figure in the United States paediatric world at the time. He qualified at Harvard Medical School in 1935. In World War II he served in the Army Medical Corps, returning to research at Harvard Medical School in 1946. May became an Associate Professor of Pediatrics at the University of Minnesota from 1947 to 1952, and subsequently held senior positions in Iowa, New York and Denver.
Charles May was succeeded as head of the Nutrition Clinic in Boston in 1947 by Harry Shwachman (of whom a great deal more later) who had worked with him before the war when May was the chief resident. Shwachman many years later recalled that “May’s interest in CF was limited because he didn’t have a laboratory…and he wrote a textbook on CF because he wanted to write down his experience before he left the clinic. He went to England with an army unit and gave a lot of lectures in London at Great Ormond Street Clinic in London. And Dr Norman who was in charge of that clinic told me that he learned all he knew about CF from Dr May. And I had been May’s right hand man for a number of years so it was logical that I take over the clinic when he left” (Fanos JH. 2008 Am J Med Genet Part A 146A:284-293).
It is likely that May gave the first lecture on CF in the UK at the Royal Society of Medicine during his time in the US army (May, 1943 below). Later he held the Chair of Pediatrics at the University of Iowa where later he chaired the first scientific meeting of the National CF Research Foundation in 1955 which was attended by most of the few US paediatricians involved in CF care. Also, around 1953, he was one of the clinicians to observe that Pseudomonas aeruginosa was usually involved when persisting pulmonary infection was present. According to Warren Warwick, May also authored a seminal work on pancreatic enzymes (not located).
1946 The first report of the dramatic effect of the new antibiotic, penicillin, in children with cystic fibrosis
1946 Di Sant’Agnese PA, Andersen DH. Celiac Syndrome IV. Chemotherapy in infections of the respiratory tract associated with cystic fibrosis of the pancreas; observations with penicillin and drugs of the sulphonamide group, with special reference to penicillin aerosol. Am J Dis Child 1946; 72:17-61.

Fig 10. Paul di Sant’Agnese
This is the first report of the use of penicillin in cystic fibrosis. In 1943 a small quantity of penicillin became available from the US Army to treat three children with CF and a further 2 in 1944 with intramuscular penicillin; the results were variable.
Bryson and co-workers at the Carnegie Foundation were first to investigate the use nebulised penicillin (Bryson et al, Science 1944; 100:33). In 1944 Alvan Barach of the Presbyterian Hospital was using a penicillin aerosol to treat asthma and bronchiectasis (Barach et al, Ann Int Med 1945; 22:485). Similar apparatus was used by di Sant ‘Agnese for these children with CF; it consisted of a rubber mask with re-breathing bag and nebuliser with a flow of oxygen into the nebuliser (figure 11).
This is an interesting first report describing early treatment in CF and in particular the first use of nebulised penicillin 20,000 units seven times daily with or without intramuscular penicillin 160,000 units daily. According to di Sant’Agnese, dramatic response occurred in 15 infants and young children with CF who eventually received the treatment but little success was achieved in infants less than a year old.
From 1947 there were numerous publications on the use of the recently available penicillin in other conditions by various routes both oral, by aerosol and even powdered inhalations – no less than 18 such publications were reviewed in The 1948 Year Book of Pediatrics. (Poncher HG (ed). Year Book Publishers. Chicago). Undoubtedly a new era of treatment of infection had begun.
Di Sant’Agnese (1914 – 2005) (figure 10) wrote that “Penicillin is the first drug known to affect the course of fibrocystic disease after cyanosis has marked the existence of suppurative Staphylococcal bronchitis”. Later, in 2001, he recalled “In most patients the results (of penicillin treatment) were dramatic. From death’s door, slowly dying from chronic pulmonary disease while we watched helplessly, patients revived in a few days”. Sulphonamides were said to be useful in prophylaxis and for intercurrent infections but not after the stage of suppurative bronchitis.
Dr Lynn Taussig, who worked with di Sant’Agnese, later recalls that Paul di Sant’Agnese and Harry Shwachman were definite rivals but had considerable respect for each other. Di Sant’Agnese came from a noble family in Italy. His father was an obstetrician and radiotherapy expert who was physician to the Italian Royal Family. Paul went to Rome medical school and then came to USA in 1939 to study medicine in New York. He was chief resident in paediatrics and published his first paper on tick paralysis – as initially he worked in infectious disease and immunology; also he studied the effect of immunisations in early life. He went to work with Dorothy Andersen at the Presbyterian Hospital in New York. In 1950 he described glycogen storage disease of the heart (Pompe’s disease) and as a gastroenterologist in the early 1950s he wrote on coeliac disease.
In 1953 di Sant’Agnes’s observation of abnormal sweat electrolytes in CF was one of the most important observations in the history of CF and provided the basis of the sweat test (Di Sant’Agnese et al, 1953 described below). He wrote over 140 papers mostly on CF – physicochemical differences, mucoproteins, calcium in secretions, and with Dr West the first report of pulmonary function tests, pneumothorax, ventilation, pancreas, duodenal contents, and measurements of absorption. He was the first to describe the liver changes – a few months before Shwachman’s group. He had an interest in every aspect of the condition. But the patient always came first and he was an outstanding clinician as well as a gifted researcher.
In 1955 di Sant’Agnese was involved with the medical aspects of the CF Foundation. In 1960 in Europe he met Archie Norman, David Lawson, and the International Cystic Fibrosis and Mucoviscidosis Association (ICFMA) had 28 delegates and guests from 14 countries. Di Sant’Agnese said he was most proud of training so many associates and colleagues in CF care and research.
One of his quotations – “If your plan is for one year, plant rice, if for 10 years plant trees, if for life – educate!”
1946 The first clear demonstration that cystic fibrosis was inheretied in a Mendelian recessive manner
1946 Andersen DH, Hodges RC. Celiac syndrome V. Genetics of cystic fibrosis of the pancreas with consideration of the etiology. Am J Dis Child 1946; 72:62-80.
Investigating 47 of their own families and 56 from the literature, the authors concluded that the familial incidence indicated a hereditary disorder with a recessive mode of inheritance, but which required more than one factor for its expression. Although the incidence in siblings approximates to the 25% expected of a Mendelian recessive trait, an hereditary condition requires more than one factor for its expression. This was a quite different explanation to that given by Fanconi et al, 1944 but in line with that of Bodian, 1952. Andersen continued to believe that “the pulmonary infection is the result of the nutritional deficiency” – obviously she considered this to be the additional factor required.
This is the first clear statement, backed by clinical evidence, that CF is inherited in a Mendelian recessive manner. Others, including David Lawson, would also speculate on the relative contribution of the basic defect and the secondary effects of the defect (such as chest infections and malnutrition) to the ultimate outlook for the patient. i.e. if the secondary effects could be prevented would the outlook be much better? Subsequent progress showed that this was indeed the case.
1951 The report that eventually resulted in Paul di Sant’Agnese searching for and eventually finding the cause of salt depletion during the 1948 New York heat wave
1951 Kessler WR, Andersen DH. Heat prostration in fibrocystic disease of the pancreas and other conditions. Pediatrics 1951; 8:648.
One of the most important papers up to that time from New York. Walter Kessler, the senior resident at the time, and Dorothy Andersen reported 12 children who had developed severe heat prostration. Ten were admitted during a New York heat wave in 1948 and no less than 7 were known to have cystic fibrosis. These were the days before air conditioning was generally available in New York and 1948 was a particularly hot summer. Paul di Sant’Agnese, was working with Dorothy Andersen at the time and looking after her patients; later said that he treated these particular infants as Andersen was away vacationing in Europe when they were admitted! The authors of this present report queried whether the sweat glands, as well as the glands of the pancreas and other organs, were inadequate in function or alternatively a low grade infection lowered the margin of tolerance to increased temperatures.
At the time of this report there was no explanation as to why infants with CF were particularly susceptible to heat prostration and salt depletion – fortunately Paul di Sant’Agnese decided to find out! This was the first report that children with CF were particularly susceptible to heat. It was this original incident that eventually led Paul di Sant’Agnese, who was working with Dorothy Andersen, to search for the reason for salt depletion in many of these CF infants and eventually to his recognising the abnormally high sweat sodium and chloride, and to a lesser extent potassium. This was undoubtedly the first and most important major advance in the understanding of the causation of CF up to that time (see di Sant’Agnese et al, 1953 below) and for some years to come.
1953 The papers of Paul di Sant’Agnese and Bob Darling reporting the raised sweat electrolytes in all people with CF – the most important discovery up to that time
1953 Darling RC, di Sant’Agnese PA, Perera GA, Andersen DH. Eletrolyte abnormalities of the sweat in fibrocystic disease of pancreas. Am J M Sc 1953; 225:67-70.
The first report of elevated sweat electrolytes in cystic fibrosis. di Sant’Agnese collaborated with Bob Darling who was the head of the Rehabilitation Department of the Columbia Presbyterian Hospital. In this department there was a constant temperature room and a method of collecting sweat. Initially two teenagers with CF and two controls were selected and although the sweating rate was similar the level of electrolytes was much higher in the patients with cystic fibrosis. Subsequently sweat from 9 CF children and 8 controls showed chloride more than three times higher in the people with CF than in the controls.This was an unexpected finding quite unrelated to any previously recognised abnormalities in the condition (di Sant’Agnese et al, 1953 below) but it was the most important observation since the clear identification of CF as a specific entity by Dorothy Andersen in 1938.
1953 di Sant’Agnese PA, Darling RC, Perera GA, Shea E. Sweat electrolyte disturbances associated with childhood pancreatic disease. Am J Med 1953; 15:777-784.
In this study there were 50 patients with CF, 9 with other pancreatic diseases and 50 controls. All the CF patients had similar elevations in the sweat electrolytes. Their adrenal and renal function was normal. The authors considered the findings to justify abandoning the term “mucoviscidosis” and returning to the unsatisfactory term “cystic fibrosis of the pancreas” until a better one was proposed.
1953 di Sant’ Agnese PA, Darling RC, Perera GA, Shea E. Abnormal electrolyte composition of the sweat in cystic fibrosis: Clinical significance and relationship to the disease. Pediatrics 1953; 12: 549-563.
This is the main paper that di Sant’Agnese himself quotes as describing the sweat electrolyte abnormality expanding on the paper of Darling et al, 1953 (above). Di Sant’Agnese mentions the original report of Kessler and Andersen 1951 (above) and also that the susceptibility of patients with CF to heat in the summers was noted also during subsequent summers after 1948.
Paul Quinton more recently recalls that di Sant’Agnese told him that the development of heat tolerance among troops sent to North Africa was attributed to adaptations to sweating, so di Sant’Agnese pursued excessive salt loss in the sweat as the most likely origin of volume depletion during the high heat stress (Quinton, 1999 below).

Fig 11. Robert Darling
Bob Darling (figure 11) was head of Rehabilitation at Columbia Presbyterian Hospital and had a constant temperature room. Di Sant’Agnese decided “as a shot in the dark to see if sweating function was impaired in CF patients that would lead to a smaller than normal volume of sweat, or whether there was something wrong with the sweat electrolyte concentration”. di Sant’Agnese continued – “In April 1952 two teenage children with CF and two controls were put in the constant temperature room and their sweat then analysed for electrolytes. To my surprise and excitement the answer was right there. There was a tremendous difference in the sweat electrolyte concentration between the two groups”. “In contrast the sweating rate was similar in the two groups”. (Described in detail by Paul di Sant’Agnese. Experiences of a Pioneer Researcher. In: Doershuk CF (ed.) Cystic Fibrosis in the 20th Century 2001; Fanos JH. 2008; 17-35.).
Sant’Agnese and colleagues showed the sweat abnormality was unrelated to renal or adrenal disease and was definitely related to sweat losses. For this present paper in Pediatrics they examined 43 people with CF, 9 patients with other pancreatic diseases and 50 controls in a room at 32.2 C for 1-2 hours. Sweat was collected onto dry gauze under adhesive waterproof plaster. Sweat chloride in the CF patients was 106 (60-160) meq/l, in controls and other pancreatic diseases only 32meq/l (4-80) and in CF the Na 133 (80-190), and in controls 59 (10-120).
This was undoubtedly the most important advance in the understanding of CF up to that time. However, astonishingly, di Sant’Agnese recalls the paper received a very cool reception at the 1953 meeting of the American Pediatric Society with not a single question! Also when presented before Jas Kuno, apparently a distinguished sweat physiologist, Kuno uttered one word -“impossible”- and walked out of the room! It is also said that even di Sant’ Agnes’s close colleague Dorothy Andersen was at first reluctant to accept the findings. However, and perhaps predictably, Harry Shwachman soon visited di Sant’Agnese in New York; he was impressed and with his usual alacrity and energy was able to present a large supportive series by October 1954 much to di Sant’Agnese’s delight!
1959 The Gibson and Cooke pilocarpine iontophoresis method of stimulating sweating was a major advance from the potentially dangerous practice of heating infants in blankets or plastic bags
1959 Gibson LE, Cooke RE. A test for concentration of electrolytes in sweat in cystic fibrosis of the pancreas utilising pilocarpine electrophoresis. Pediatrics 1959; 23:545-549. [PubMed]

Fig 12. Dr. Leoni Shapiro performing a pilocarpine iontophoresis sweat test
This classic paper by Lewis Gibson and Robert E Cook described the pilocarpine iontophoresis method for stimulating sweating to obtain sweat for analysis. It was a major advance, as prior to this these frail infants were often heated in blankets or plastic bags to obtain sweat for analysis. The pilocarpine iontophoresis method is still used by most laboratories for stimulating localised sweating (figure 12). Electrodes are applied to the infant’s forearm as pilocarpine is iontophoresed into the skin: later the sweat is collected from the grated area on weighed gauze and the weight and electrolyte content estimated in the laboratory.
It is important that an experienced biochemist carries out the test. Subsequently, as more laboratories performed the test caution was expressed as when performed by inexperienced operators mistakes occurred. These mistakes often resulted in CF being over diagnosed when the sweat result was falsely high which could happen if the sweat specimen was allowed to evaporate. Also, there were reports of burns under the electrodes.
The first report of such major diagnostic errors in the UK was from Birmingham (Smalley et al, Lancet 1978; ii: 415-417 below) from Prof. Charlotte Anderson’s unit but soon others followed (David TJ & Phillips BM. Lancet 1982; ii: 1204-1205 [PubMed]). In Leeds we found that 7 (4%) of the first 179 children referred for Comprehensive Assessment of their CF from various parts of the UK did not have the condition (Shaw NJ & Littlewood JM. Arch [PubMed]
The consequences of a false diagnosis of CF frequently had disastrous socio-medical consequences e.g. one mother had been sterilised to avoid having another child with CF as it was mistakenly thought her first child had CF. Some parents had avoided having further children; some parents took legal action against the paediatrician involved in the mistaken diagnosis.
So the sweat test is an excellent test if carried out correctly by experienced laboratory staff but can be disastrous if performed by inexperienced staff as an occasional procedure. Also the result should not be accepted uncritically by the paediatrician but should be considered with all the other clinical and

Fig 13. Lewis Gibson
laboratory evidence when making a diagnosis of cystic fibrosis.
Dr Lewis E Gibson (1928-2008) was a paedaitrician and researcher who developed the pilocarpine iontphoresis sweat test after he moved to the NIH in 1955. He recalls seeing his physiology teacher raising a hive on his own arm by iontophoresing histamine into the skin with an electric current. The difficulties in obtaining sweat by heating (“a few babies had died in sweat bags”) stimulated his further interest in iontphoresis of pilocarpine to stimulate sweating. in 1957 Gibson returned to Johns Hopkins where the Chairman of Pediatrics, Dr Robert E Cooke, had worked with CF and had some experience of iontophoresis. They combined on the present publication which apprently became a Current Contents classic. There is an interesting chapter by Dr Gibson in Dr Carl Doeshuk’s book (Cystic Fibrosis in the 20th Century. Ed.Doershuk CF. AM Publishing Ltd Cleveland, Ohio. 2002). Dr Robert E Cooke (figure 13) was Chairman of the Department of Pediatrics at Johns Hopkins from 1956 to 1973 and involved in many areas of paediatrics and child health other than CF on both a local and national leve; these included programmes that brought better care to inner city mothers and their children, the Kennedy Program for Research into the Etiology of Mental Retardation and eventually as Director of the Kennedy Institute for Handicapped Children.
1964 CF Centre care by a team of professionals who gain experience in treating many patients is regarded as the best way of providing hospital care for people with CF. The method was pioneered by Leroy Matthews and Carl Doershuk in Cleveland and described in these papers
1964 Matthews LW, Doershuk CF, Wise M, Eddy G, Nudelman H, Spector S. A therapeutic regimen for patients with cystic fibrosis. J Pediatr 1964; 65:558-575. [PubMed]

Fig 15. Leroy Matthews

Fig 14. Carl Doershuk
Dr Doershuk (figure 14) recalls that Dr William Wallace, Chairman of Paediatrics at the Babies and Children’s Hospital, Cleveland had been approached in 1957 by a parents’ organisation – the “Cousins Club” – one of whom had already lost a child with CF and had another deteriorating from the condition. They asked Dr Wallace to start a “research orientated treatment programme for CF” which they offered to fund.
To develop this programme Dr Wallace appointed a young paediatrician, Dr Leroy Matthews (figure 15) to plan and initiate the “comprehensive and prophylactic (preventive) treatment programme” for the treatment of cystic fibrosis. The programme which developed eventually became the model for the CF Foundation CF centres programme (also Doershuk et al, 1964 & 1965 below).
Three important areas of treatment were the obstructive pulmonary lesion, the secondary infections and the pancreatic deficiency and nutritional state. Treatment was early and comprehensive even started before symptoms – where this group differed from others and as such were ahead of their time. This paper describes the “comprehensive therapeutic regimen” which so influenced CF care in N America.
Veteran CF physician Dr Warren Warwick of Minnesota has “fond memories of two great stars – Harry Shwachman and Leroy Matthews”. Of Leroy Matthews he writes – “Leroy Matthews, the greatest genius CF has seen, single handedly established the value of Comprehensive Treatment, laid the ground work for Pediatric Pulmonology, organised and led the CF Centres as well as planning and directing excellent research. He made only two mistakes. He allowed his “Comprehensive Treatment” plan to be equated with “mist tent therapy” so when the mist tent was discredited many also felt the Comprehensive Care Program was discredited. And he tried too hard to control his diabetes and suffered hypoglycaemic brain injury and cardiovascular complications” (Warren Warwick in Doershuk CF (ed). Cystic fibrosis in the 20th Century. AM Publishing, Cleveland 2001:319)
1964 Doershuk CF, Matthews LW, Tucker AS, Nudelman H, Eddy G, Wise M, Spector S. A 5 year clinical evaluation of a therapeutic program for patients with cystic fibrosis. J Pediatr 1964; 65:677-93. [PubMed]
A detailed evaluation of the results of the Cleveland comprehensive therapeutic regimen. 96 consecutive patients were followed for 18 to 60 months (average 37 months) and evaluated using a modified Shwachman score. 82% improved, 11% remained the same, and 4% showed progression beyond their initial status and only 3% died – none were less than five years of age. Patients who were regarded as having reversible pulmonary changes were reviewed separately in 1965 (Doershuk et al, Pediatrics 1965; 36:675 below).
1965 Doershuk CF, Matthews LW, Tucker A, Spector S. Evaluation of a prophylactic and therapeutic program for patients with cystic fibrosis. Pediatrics 1965; 36:675-688. [PubMed]
Good results were reported in the group of children treated prophylactically with little progression over an average of 4.5 yrs. The early intervention and prophylactic approach was not the usual policy at this time and most clinicians waited until symptoms developed – even experts such as Paul di Sant’Agnese. In this study 98 consecutive patients had been followed for an average of 4.5 years and the clinical course of 49 were considered to be on prophylactic therapy was significantly different from the accepted natural course of the disease and from the 49 patients who had irreversible lung damage when first seen. No evidence of significant progression of the pulmonary state was seen in any of the prophylactic group. No deaths occurred in this group and the annual mortality rate was only 2% for the whole group. Their findings supported the need for early diagnosis and prophylactic treatment
Dr Sydney Gellis (always a sceptic regarding the treatment of CF!!) in the Year Book of Pediatrics questioned whether more mild cases had been included; also whether the improved survival could not have been due entirely to antibiotics and to none of the other methods of treatment described such as mist tents, aerosols, segmental postural drainage. di Sant’Agnese also observed that in one series of older patients (Shwachman et al, 1965) the diagnosis had not been made in many until teen age years suggesting they had a milder form of CF.Certainly patients with CF diagnosed later in childhood, who were included in the early adult series, undoubtedly more frequently would have had milder CF gene mutations as was confirmed in later studies (Gan K-H et al, 1995 below). Rather surprisingly di Sant’Agnese questioned the need to start the full prophylactic programme in all patients as soon as the diagnosis is made and agrees with most other clinicians at that time that treatment is not started “until there is indication of incipient pulmonary involvement”. This view regarding the start of treatment is interesting and in these days of neonatal screening, failure to start early microbiological monitoring, early eradication treatment of respiratory pathogens and early nutritional intervention, would be regarded as quite unacceptable.
1974. Douglas Crozier’s abandonment of dietary fat restriction and introduction of more intensive treatment results in improved nutrition, growth and survival
1974 Crozier DN. Cystic fibrosis: a not so fatal disease. Pediatr Clin North Am 1974; 21:935-948. [PubMed]

Fig 16. Douglas Crozier giving the “Breath of Life Award ” to Maureen McChesney in February 1970 (reproduced with family’s permission)
This paper gives an idea of treatment at the Toronto CF clinic in the early Seventies. Dr Douglas Crozier (figure 15a), who started the Toronto CF clinic in 1958, stated that “success of treatment will depend on a complete assessment of the patient and then continuing attempts to obtain normal bodily function and maintain it”. He described how he advised his patients to abandon the traditional low fat diet and used of very high doses of pancreatic enzymes (up to 100 Cotazym capsules per day). Crozier believed that “to deprive a child with cystic fibrosis, who usually has very little subcutaneous fat, of this important nutrient seems ridiculous”.
The superior nutritional state of the Toronto patients is believed to be the main reason for their better survival. In 1973, 428 people with CF were attending the Toronto clinic of whom 92 (21.4%) were 16 years or older – this was quite remarkable for that time. The nutritional management at the Toronto clinic was considered to be the main factor in the superior survival figures of the Toronto patients (Corey M et al. J Clin Epidemiol 1988; 41:588-591).
Although I was never fortunate enough to meet Crozier I was profoundly influenced by this landmark paper from Toronto regarding the approach to management of people with CF – also by a later visit to Crozier’s successor, Henry Levison, at the Toronto clinic and attendance at the 8th International Cystic Fibrosis Congress there in 1980.
On the basis of Crozier’s recommendation to “perform a complete assessment of the patient” and then “continuing attempts to obtain normal bodily function and maintain it”, in 1980, on returning to Leeds from the Toronto meeting, with the help of a few colleagues at St James’s in Leeds, we started regular Comprehensive CF Assessments on all our children with cystic fibrosis and those referred to me from our region. The advice based on the results of a Comprehensive Assessment proved to be a major factor in the development of the Leeds Regional CF referral service.
1977 The first report of the acid resistant pancreatic enzyme Pancrease – this and subsequent acid resistant preparations revolutionised nutritional management during the Eighties
1977 Khaw KT, Adeniyi-Jones S, Gordon D, Polombo J, Suskind R. Efficacy of pancreatin preparations on fat and nitrogen absorptions in cystic fibrosis. Pediatr Res 1978; 12:437.

Fig 18. Pancrease microspheres still intact in aspirated duodenal fluid

Fig 17. Creon (top) and Pancrease microspheres
An early report of the marked superiority of Pancrease over currently used enzymes at the time. Pancrease consisted of acid resistant micro spheres which only released their enzymes when they reached the more alkaline environment of the upper intestine, thus protecting their active enzyme contents from destruction by the gastric acid (figure 18).
Viokase and Cotazym were the standard enzyme preparations in use at the time both of which were affected by gastric acid. In this first report twelve children with CF aged eight to 14 years took Viokase four eight or 12 tablets, Cotazym two four or six capsules and Pancrease one two or three capsules per meal. All three preparations improved absorption compared with placebo but Pancrease did so at the much smaller dose. Viokase 12 capsules per meal and Pancrease 3 capsules per meal achieved 94% and 94.8 % fat absorptiion respectively. Similarly six Cotazym and three Pancrease capsules achieved 84.2% and 89.7% fat absorption respectively. The acid resistant microspheres, particularly first Pancrease and soon after Creon, were undoubtedly one of the major nutritional advances, when gradually introduced during the early Eighties allowing most patients to take a normal fat intake and hence significantly improve their energy intake. The markedly better absorption of fat and protein was shown in all subsequent trials and the reduction in the patients’ unpleasant bowel symptoms and fat intolerance in many was quite dramatic (also Weber et al, 1979 below).
1979 Description of the immunoreactive trypsin test for newborn screening – eventually used in neonatal screening programmes worldwide
1979 Crossley JR, Elliott RB, Smith PA. Dried blood spot screening for cystic fibrosis in the newborn. Lancet 1979; i: 472-474. [PubMed]

Fig 19. Jeanette Crossley
This study was from Auckland, New Zealand. Serum-immunoreactive-trypsin (IRT) was measured in children with CF and a variety of controls. In the first few months of life all the children with CF had a raised serum-IRT. A dried blood-spot assay for IRT was established using double antibody radioimmunoassay kit (Hoechst Research Laboratories) and presented as having potential as a screening test for CF in the newborn. There were none of the disadvantages of stool trypsin screening as infants with residual pancreatic function (“pancreatic sufficient” infants) also had a raised IRT
In New Zealand Jeanette Crossley (figure 19) and colleagues further evaluated the test (Crossley JR et al. Clin Chim Acta 1981; 113:111-121.[PubMed]; Lyon IC et al. NZ Med J 1983; 96:673-675). Neonatal IRT screening was soon adopted in New Zealand and some states in Australia where Bridget Wilcken of Sydney has been a champion of neonatal screening since the early Eighties. The IRT test now forms the basis of most neonatal CF screening programmes throughout the world – now usually combined with DNA examination for CF-causing mutations when the IRT is positive.This was a really important landmark paper for neonatal CF screening. Neonatal IRT screening had the great practical advantage of using the same neonatal blood spots already collected by the heel prick in the first week of life for phenylketonuria and later hypothyroidism.

Fig 20. Anthony Heeley
In the UK, Dr Anthony Heeley (figure 20), the biochemist in Peterborough, UK started IRT neonatal screening in East Anglia in 1981 (Heeley et al, 1982 below). In 2009 he kindly commented on the early stages of IRT screening as follows –
“Your summaries of our early papers are excellent. A point to emphasise was that Crossley & Elliott’s seminal work in 1979 required 2 x 12 mm blood spots for the assay. We realised that was unrealistic for routine screening and in our paper in the Lancet the same year (King DN. Heeley AF. Walsh MP. Kuzemko JA. Sensitive trypsin assay for dried-blood specimens as a screening procedure for early detection of cystic fibrosis. Lancet 1979; 2(8154):1217-1219) we showed that their results could be confirmed using 4 mm blood spots which was a much more practical size for routine prospective screening trials. These trials were quickly underway in the Antipodes, here and elsewhere, but we have ascertained that the first case of CF to be detected in a prospective screening trial with IRT was in East Anglia in 1980, a girl born one month before the first case detected prospectively by the New Zealanders. Even better, although homozygous Delta F 508, to my knowledge she led a robust and active life up to late teenage when I lost contact. Her paediatrician could not recall any exacerbation of her condition requiring hospital admission. I still treasure a slide of her as a healthy teenager and I was proudly able to show it at a National Neonatal Screening Seminar in Newcastle a couple of years ago (dragged out of retirement for the historical story!)”
.In 2009 Jeanette Crossley kindly commented for me as follows –
“We were fortunate that our 1979 Lancet paper robustly established the potential of blood spot IRT for CF newborn screening, using the Hoechst-Behring Riagnost Trypsin Kit. The kitset as supplied required a relatively large sample size [we used 2 x 12mm discs from a 5-day Guthrie card], so our next step was modifying that method. We achieved 10-fold greater sensitivity, enabling one 3mm disc as sample, left in the tube throughout the stages of the assay. In a retrospective study of 23 known CFs using this optimized assay [unpublished], each CF was clearly distinguishable from two controls from the same batch of Guthrie cards, despite the Guthrie cards having been stored at room temperature for up to seven years.
For several reasons, economics being a main one, we did not conduct an extensive prospective trial using the Hoechst-Behring reagents. Rather, we developed our own Auckland radioimmunoassay ‘from scratch’ [reported in detail in the Clin Chim Acta paper1981]. Its validation included repeating the retrospective study, again clearly distinguishing all the known CFs from controls. We established that the molecular species assayed in blood is trypsinogen. Our ‘Auckland assay’ was more sensitive, and had better linearity for serially diluted samples, than our optimization of the Hoechst-Behring kitset assay. Also, knowing what was in all our reagents gave us confidence – we had been unable to obtain from Hoechst the exact make-up of their assay.
Our team effort launched dried bloodspot CF screening in NZ, first as a pilot, and then nationally – and also enabled Bridget Wilcken to get started in NSW, Australia. I’ve been told that our original purified trypsin and antisera were used in the National CF Screening Programme until about 1990, when changing health and safety policies required a move away from radio-labelled methods”.It had been a delight to be invited to present our experience with our ‘Auckland IRT assay’ and with our optimisation of the Hoechst-Behring Riagnost kitset, at an international meeting ‘Immunoreactive Trypsine’ sponsored by Hoechst France in Caen, 25 October 1980. We were very pleased to see the quick and thorough progress being made in several countries with validation of IRT as a CF newborn screening method. The first UK symposium on neonatal screening for cystic fibrosis, for example, sponsored by CIS (UK) Ltd and Sorin Biomedica SpA, was held in London 10 days after the Caen meeting”.
1981 The first major advance in understanding the basic defect since the discovery of the abnormal sweat electrolytes by Sant’Agnese in 1953
1981 Knowles MR, Gatzy JT, Boucher RC. Increased bioelectric potential difference across respiratory epithelia in cystic fibrosis. N Eng J Med 1981; 305:1489-1495. [PubMed]

Fig 21. Richard Boucher

Fig 21. Michael Knowles
Undoubtedly this was a major landmark paper. Michael Knowles from North Carolina describes how they developed a technique that measured a single parameter of epithelial function – the transepithelial potential difference (PD); this they used to define salt (ion) transport properties of nasal and lower respiratory epithelium in normal humans in vivo and then in patients with CF whose transepithelial PD they showed was markedly higher than normal – a feature apparently present within hours of birth suggesting a primary genetic epithelial defect rather than due to any circulating “CF factor” or effect from infection.
Knowles and colleagues eventually identified a defect in the ability of Cl to move across CF cells, the Cl permeability of the airway epithelium was not activated by beta-agonists and there was a very rapid absorption of salt and water. This led to the theory that CF airway epithelium has an excessive rate of NaCl transport and water absorption leading to dehydration of the secretions, reduced clearance of mucus predisposing to chronic airway infection – a theory which increased in popularity and was championed particularly by Knowles’s colleague Richard Boucher.
Professor Michael Knowles (figure 21) was honoured with an award from the CF Foundation at the 2008 Annual North American CF Conference. He and Richard Boucher are certainly two of the outstanding North American CF clinicians and researchers of the era.
1981 The start of widespread use of inhaled antibiotics in the UK to stabilise patients with chronic Pseudomonas aeruginosa respiratory infection
1981 Hodson ME, Penketh ARL, Batten JC. Aerosol carbenicillin and gentamicin treatment of Pseudomonas aeruginosa in patients with cystic fibrosis. Lancet 1981; i: 1137-1139. [PubMed]
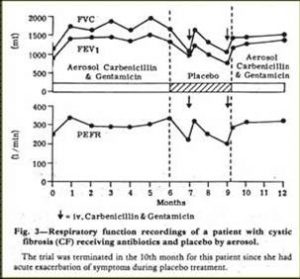
Fig 23. Stabilisation of CF patient with inhaled antibiotics. Hodson et al.1981

Fig 22. Margaret Hodson
Another definite landmark paper from the Adult CF Unit at the Royal Brompton Hospital, London. by Dr Margaret Hodson (figure 22), later Professor of Cystic Fibrosis there. Professor Hodson was to make many major contributions to the treatment of people with CF from her vast experience at the Brompton Hospital treating many hundreds of adult patients with CF. In 2012 she was awarded the OBE for services to medicine.
This early paper of Margaret’s had a major influence on treatment in the UK although there had been many earlier concerns about increasing the incidence of bacterial resistance from nebulised antibiotics. Although nebulised penicillin had been used in the Forties when S. aureus was the main pathogen (di Sant’Agnese, et al, 1946 above) it was undoubtedly this present paper that revived the interest in nebulised antibiotics for patients with chronic Pseudomonas infection – particularly in the UK. The nebulised anti-Pseudomonal antibiotics obviously stabilised the condition of some patients with relapsing chronic P. aeruginosa infection who were requiring increasingly frequent courses of IV antibiotics and represented a major milestone in treatment.As a result of this paper nebulised anti-Pseudomonal antibiotics became widely used in the UK for patients with chronic Pseudomonas infection. In this respect the UK was well in advance of North America where, even in 1986, McLusky and colleagues from Toronto advised that, “until additional well-controlled trials were completed their routine use (of inhaled antibiotics) was not justified because of cost, potential side effects and the propensity to select resistant organisms” (McLusky et al, 1986 below). As it turned out, both the routine use of anti-Pseudomonal antibiotics to suppress chronic infection as recommended by Margaret Hodson in 1981 (Ramsay BW et al, 1999) and the early use of colomycin to eradicate early infection (Littlewood et al, 1985; Valerius et al, 1991) both proved to be effective, proven and eventually widely used treatments for people with CF on both sides of the Atlantic.
1983 Improved survival with regular courses of intravenous anti-Pseudomonal antibiotics in chronically infected patients in Copenhagen – at that time a new concept in aggressive treatment which influenced many clinicians
1983 Szaff M, Hoiby N, Flensborg EW. Frequent antibiotic therapy improves survival of cystic fibrosis patients with chronic Pseudomonas aeruginosa infection. Acta Paediatr Scand 1983; 72; 651-657. [PubMed]

Fig 23. Erhard Flensborg

Fig 24.Niels Hoiby
This is one of the main reports of improved survival at the Danish CF Centre since starting 3-monthly courses of intravenous anti-Pseudomonal antibiotics for chronically infected patients from 1976. During the period 1971-75, 51 chronically infected CF patients were treated with IV antibiotics but only when their condition deteriorated. During the period 1976-80, 58 chronically infected CF patients were treated with regular courses of intravenous antibiotics every three months.
The five year survival of patients from the time of the onset of their chronic P. aeruginosa infection increased from 54% in the first period (1971-1975) to 82% in the second period (1976-1980). The authors concluded that intensive “maintenance” chemotherapy against P. aeruginosa improves survival and quality of life of CF patients although permanent eradication of chronic P. aeruginosa is not accomplished. These results were largely ignored outside Denmark even in Europe. True, this was not a controlled trial and there were problems with the cost of the antibiotics, drug allergies, the patients’ time and quality of life; also there were other treatments which could have favourably affected the outcome. It is important to note and particularly relevant that long term nebulised antibiotics, that frequently stabilise the respiratory function of chronically infected patients, were not used at the time of this study. However, it is a measure of how far ahead of many clinics the staff at the Copenhagen centre were as many clinicians were still using IV antibiotics only as a last resort or not at all (as in the case of some children referred to our Leeds CF centre for Comprehensive Assessment even in the early Eighties). Unfortunately, even in 2018, there is still no totally adequate controlled trial of this form of regular IV antibiotic treatment although many clinicians have increasingly adopted a policy of very early intervention – which in practice almost amounts to 3-monthly treatments for some patients. One study from the UK comparing 3-monthly elective with symptomatic treatment showed no advantage in elective treatment but even the “treat when symptomatic” group received 3 courses of IV antibiotics annually (Elborn et al, Thorax 2000; 55:355-358 below).

Fig 25. Niels receiving the ECFS Award in 2012 from the then president Stuart Elborn
Perhaps the frequency of intravenous antibiotic treatments is best left to the judgement of the clinician and the CF team

Fig 26. After the 25 year celebration at Copenhagen University with Niels and Hanne Wendel Tybkjaer
who know the patient – and last, but not least, to the patient’s opinion! Certainly the availability of effective inhaled antibiotic therapy has had a major influence on the management of the patients with chronic P. aeruginosa infection and reduced the need for intravenous antibiotic treatments in many patients.
Professor Niels Hoiby received the ECFS Award in 2012 (figure 25) in recognition of his quite outstanding contribution to the understanding of Pseudomonas lung infection and translating this knowledge into better outcomes for people with cystic fibrosis. He was also recognised for his contribution as the Founding President of the European Cystic Fibrosis Society and his lifelong commitment to the CF center in Copenhagen.
In 2013 Niels celebrated his 25 years as Professor of Microbiology in Copenhagen University. I was privileged to be invited to reflect on his major and quite unique contribution to our understanding and treatment of cystic fibrosis at this 25th Anniversary Meeting at Copenhagen University (figure 26). Hanne Wendel Tybkjaer, previously CEO of the Danish CF Association, now works with CF Worldwide.
1983 Another major scientific milestone – ion impermeability rather than defective ion exchange
1983 Quinton PM. Chloride impermeability in cystic fibrosis. Nature 1983; 301:421-422. [PubMed]

Fig 26. Paul Quinton
Another landmark paper in the understanding the CF defect by Professor Paul Quinton (figure 26). He later recalls (Quinton, 1999 below) that Mike Knowles (1981 above) reported a significantly larger than normal electronegative potential across the nasal epithelium, along with the fact that NaCl absorption was inhibited in the CF sweat ducts and also that sodium was relatively more absorbed than chloride. This gave Quinton the idea that the basic defect in the CF duct had to be due to an anion impermeability and not defective anion exchange. Using a series of microperfusion experiments of sweat glands (it is said some from his own forearms) Quinton measured the electrolytes by “energy dispersive X-ray analysis”. The chloride impermeability he had shown in sweat glands was the basis for the raised sweat electrolytes and provided a physiological explanation for the high salt content of CF sweat and also “provided the first description of a basic cellular defect that has since proven to be uniformly inherent in all CF affected cells”.
Paul Quinton (figure 26) holds the Nancy Olmsted Chair of Pediatric Pulmonology UC San Diego and is Professor of Biomedical Sciences at UC, Riverside. There is an very interesting interview with Paul Quinton on the Heroes of Hope website (http://www.heroesof hope.comnov09.jsp)
1985 The first heart-lung transplants in CF patients by Mr Yacoub’s team in London provided hope for those with terminal cystic fibrosis.
1985 (Published in 1990) Yacoub MH, Banner NR, Khaghani A, FitzGerald M, Madden B, Tsang V, Hodson M. Heart-lung transplantation for cystic fibrosis and subsequent domino heart transplantation. J Heart Transplant 1990; 9:459-467.

Fig 27. Sir Magdi Yacoub
A major advance, for those who had reached the end stages of their disease (an FEV1 predicted of less than 30%), was the

Fig 28. John Wallwork
successfulintroduction of heart-lung transplantation in 1985, pioneered by Sir Magdi Yacoub (figure 27) and his team in London (Yacoub et al, 1990; Scott et al, 1988) and Mr John Wallwork (figure 28) and his team in Cambridge. The possibility of successful treatment in what were previously the terminal stages of the condition had a major influence on both prognosis and the treatment of severely affected individuals.
The first results of heart-lung transplantations were quite remarkable and were related both to surgical skills, concentrated medical expertise in assessment and after care and also to more successful immunosuppressive therapy to prevent rejection of the transplanted organs. Later double lung transplants became more popular (Pasque et al, 1990 below) and in recent years are now the favoured operation. Living donor lung transplants have proved successful in some centres (Cohen & Starnes, 2001 below; Mohite PN et al, 2013).

Fig 30 Bruce Reitz

Fig 29. Norman Shumway
Dr Norman Shumway (1923-2006) (figure 29) laid the groundwork for heart lung transplantation with his experiments into heart transplantation at Stanford in the mid-1960s. Shumway performed the first adult heart transplant in the US in 1968. Building on his research at Stanford, Dr.Bruce Reitz (figure 30) performed the first successful heart–lung transplant on Mary Gohlke in 1981 at Stanford Hospital. The transplant team at Stanford is the longest continuously active team performing these transpla
1985 Two papers that resulted in localisation of the CF gene to chromosome 7 and had a major influence on the search for the gene
1985 Eiberg H, Mohr J, Schmiegelow K, Neilsen LS. Linkage relationships of paraoxinase (PON) with other markers: evidence of PON-cystic fibrosis synteny? Clin Genet 1985; 28:265-271. [PubMed]
This was the first positive move towards narrowing down the identification of the CF gene. The linkage relationships of the serum arylesterase paraoxonase

Fig 31. Hans Eiberg
(PON) was examined in normal Danish families and in Danish and English CF families. The highest correlation was found between the inheritance of paraoxinase and cystic fibrosis. Linkage studies for PON against 64 other polymorphic marker systems did not give such a close relationship. By the present screening about 2/3 of the genome could tentatively be excluded as the region of PON and cystic fibrosis. (Synteny = the presence together on the same chromosome of two or more gene loci).
Prof. Hans Eiberg (figure 31) is a Danish geneticist, known for the discovery of the genetic mutation causing blue eyes. Together with professor Jan Mohr he established Copenhagen Family Bank in 1972, a store of DNA samples, comprising about 1000 large Danish families. Hans Eiberg has contributed to the mapping of the human genome, and he has succeeded in finding important genetic markers for several serious illnesses in addition to cystic fibrosis.
1985 Tsui L, Buchwald M, Barker D, Braman JC, Knowlton R, Schumm JW, Eiberg H, Mohr J, Kennedy D, Plavsic N, et al. Cystic fibrosis locus defined by a genetically linked polymorphic DNA marker. Science 1985; 230:1054-1057. [PubMed]

Fig 32. Lap Chi Tsui
Professor Lap Chi Tsui (figure 32) and co-workers used a restriction fragment to localise the gene to the long arm of chromosome 7. In a set of 39 families, a polymorphic DNA marker was genetically linked to the autosomal recessive gene that causes cystic fibrosis. The DNA marker (called D0CRI-917) was also linked to the paraoxinase locus, which by independent evidence is linked to the CF locus (Eiberg et al, 1985 above). The location of the CF gene was now narrowed to about 1 percent of the human genome (about 30 million base pairs). This was the first step in molecular analysis of the CF gene. This was a major step forward localising the CF gene to chromosome 7. Apparently Lap Chi Tsui encountered several problems when working with the pharmaceutical firm, Collaborative Research Inc., that had already invested $10 million in the project; they could see the commercial opportunities of developing markers for antenatal and carrier diagnosis.
The problems are well described by Leslie Roberts (Gene transfer test back on track. Science 1988; 240:141-144 & 282-285). Essentially Collaborative Research Inc. wanted to delay publication of the location of the gene on chromosome 7 until more markers could be identified and patents could be applied for. Meanwhile other groups, Williamson’s in London (Wainwright et al, 1985 below) and Ray White’s in Utah (White et al, 1985 below) were said to be aware of the findings that the CF gene was on chromosome 7 and had identified other markers very close to the CF gene.
1989 The three papers that appeared in the same issue of Science in 1989 describing the identification of the CF gene
1989 Kerem B-S, Rommens JM, Buchanan JA, Markiewicz D. Cox TK. Chakravarti A. Buchwald M. Tsui LC. Identification of the cystic fibrosis gene: genetic analysis. Science 1989; 245:1073-1080. [PubMed]

Fig 33. Batsheva Kerem
Approximately 70 percent of the mutations in cystic fibrosis patients correspond to a specific deletion of three base pairs, which results in the loss of a phenylalanine residue at amino acid position 508 of the putative product of the cystic fibrosis gene. Extended haplotype data based on DNA markers closely linked to the putative disease gene locus suggest that the remainder of the cystic fibrosis mutant gene pool consists of multiple, different mutations. A small set of these latter mutant alleles (about 8 percent) may confer residual pancreatic exocrine function in a subgroup of patients who are pancreatic sufficient. The ability to detect mutations in the cystic fibrosis gene at the DNA level had important implications for genetic diagnosis.

Fig 34. Jack Riordan
1989 Riordan JR, Rommens JM, Kerem B-S, Alon N. Rozmahel R. Grzelczak Z. Zielenski J. Lok S. Plavsic N.Chou JL. et al. Identification of the cystic fibrosis gene: cloning and characterization of the complementary DNA. Science 1989; 245:1066-1073. [PubMed]
Overlapping complementary DNA clones were isolated from epithelial cell libraries with a genomic DNA segment containing a portion of the putative CF locus, which is on chromosome 7. Transcripts, approximately 6500 nucleotides in size, were detectable in the tissues affected in patients with CF. The predicted protein consists of two similar motifs, each with (i) a domain having properties consistent with membrane association and (ii) a domain believed to be involved in ATP (adenosine triphosphate) binding. A deletion of three base pairs that results in the omission of a phenylalanine residue at the center of the first predicted nucleotide-binding domain was detected in CF patients.
1989 Rommens JM, Iannuzzi MC, Kerem B-S, Alon N. Rozmahel R. Grzelczak Z. Zielenski J. Lok S. Plavsic N. Chou JL. et al. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science 1989; 245:1059-1065. [PubMed]
An understanding of the basic defect in the inherited disorder cystic fibrosis requires cloning of the cystic fibrosis gene and definition of its protein product. In the absence of direct functional information, chromosomal map position is a guide for locating the gene. Chromosome walking and jumping and complementary DNA hybridization were used to isolate DNA sequences, encompassing more than 500,000 base pairs, from the cystic fibrosis region on the long arm of human chromosome 7. Several transcribed sequences and conserved segments were identified in this cloned region. One of these corresponds to the cystic fibrosis gene and spans approximately 250,000 base pairs of genomic DNA.

Fig 36. Francis Collins

Fig 35. 1989 presentation of the Paul di Sant’Agnese Award to the leaders of the scientific teams (in italic type) – Left to right Lap Chi Tsui, Paul di SantAgnese, Evelyn Graub, Milton Graub (past President CFF), Francis Collins and Jack Riordan (Figure from Doershuk CF 2002 with permission).
1990 rhDNase (Pulmozyme) had a major effect on the treatment of many people with CF – undoubtedly one of the major clinical advances of the decade
1990 Shak S, Capon DJ, Hellmiss R, Marsters SA, Baker CL. Recombinant human DNase 1 reduces the viscosity of cystic fibrosis sputum. Proc Natl Acad Sci 1990; 87:9188-9192.[PubMed]

Fig 37. Steve Shak
An early report of the significant effect of recombinant human DNase1 (Pulmozyme) on sputum viscosity in people with CF by Steve Shak (Figure 37). To evaluate the potential clinical utility of recombinant human DNase I (rhDNase) in the treatment of CF, the authors cloned, sequenced, and expressed rhDNase. Catalytic amounts of rhDNase greatly reduced the viscosity of purulent CF sputum, transforming it within minutes from a non-flowing viscous gel to a flowing liquid. The reduction in viscosity is associated with a decrease in size of the DNA in the sputum.

Fig 38. Henry Fuchs
The authors suggested that inhalation of an rhDNase aerosol may be a simple direct approach that would help individuals with CF, and other patients with pneumonia or bronchitis, to clear their airways of purulent secretions. The defiinitive Phase III trial was published in 1994 – (Fuchs HJ et al. Effect of aerosolized recombinant human DNase on exacerbations of respiratory symptoms and on pulmonary function in patients with cystic fibrosis. The Pulmozyme Study Group. N Eng J Med 1994; 331:637-642).(figure 38)
Pulmozyme was undoubtedly one of the major clinical advances of the decade and was licensed by the FDA in 1994 following this study. Regular treatment with DNase would become widely used (by over 70% of people with CF on the CFF registry by 2010) first amongst those with significant chest involvement and then later for those with milder chest problems (Quann et al, 2001 below) and eventually, in some CF units for infants. The cost (approximately £7000 per year in the UK) proved a problem in certain parts of the UK.
It is of interest that as far back as 1950 Armstrong JB & White JC (Lancet 1950; 2:739-742). [PubMed] had shown that bovine pancreatic DNase1, added to viscid purulent sputum, destroyed the extra cellular fibres of DNA and reduced the viscosity. Later Elmes PC & Armstrong JB (Thorax 1953; 8:295-300). [PubMed] reported its use in chronic bronchitis, but the side effects of the bovine preparation eventually precluded its use and further development at that time (Raskin P. Am Rev Respir Dis 1968; 98:697-698). [PubMed]
1991 The possibility of eradicating early Pseudomonas infection, first suggested in a letter to the Lancet from Leeds (Littlewood et al, 1985), was confirmed in this first Copenhagen clinical trial of early eradication therapy for Pseudomonas
1991 Valerius NH, Koch C, Hoiby N. Prevention of chronic Pseudomonas aeruginosa infection in cystic fibrosis by early treatment. Lancet 1991; 338:725-726.[PubMed]

Fig 39. Niels Henrik Valerius
A randomised controlled trial from Copenhagen confirming that early P. aeruginosa infection could be eradicated in 80% of patients with CF by three weeks treatment with oral ciprofloxacin and nebulised colistin. The infection became chronic in only 2 of 14 (14%) of treated patients but in 7 of 12 (58%) of the controls. The authors comment – “Our results thus confirm and extend the preliminary report by Littlewood et al, colleagues (1995 above) who used colistin inhalations Since chronic colonisation with Ps aeruginosa is associated with increased morbidity and mortality we recommend the use of anti-Pseudomonas treatment whenever Ps aeruginosa is isolated from the sputum of cystic fibrosis patients”
In 2012 Dr Valerius (figure 39) kindly provided this image and also commented as follows –
“Your comments on our paper are very flattering. It is the only paper on CF I ever published. my main interest having been infectious diseases and immunology, in biarticular neutrophil function. I had very close collaboration with Dr Christian Koch on this (he was in many ways my mentor, also long before he became involved in CF). I was involved in clinical work with CF in 3 periods of my professional life, from 1971-1973 as resident which was my first appointment in paediatrics and again through 1980 after returning from a research fellowship at Harvard Medical School. I wrote from Boston to Dr EW Flensborg and asked if he had a position when I would be back and he wrote back I should just say when, which was very generous indeed. Finally from 1985 until 1988, when I was appointed consultant and clinical lecturer at Department of Paediatrics at Copenhagen University Hospital at Hvidovre being mainly involved in paediatric HIV until I retired 2 years ago in 2010.I presented the paper first as a poster at the international CF Conference in Washington in 2000. It is kind of ironic that I think this poster was by far the least studied by the participants at the conference. As I recall none at all stopped by to read or discuss it – the only reaction I remember was head shaking! No bitterness now whatsoever – it’s kind of fun now and it was then as I knew I had left CF forever. But I was very surprised when the Lancet accepted it – well you always get surprised when the Lancet or NEJM accept something!”
Finally Dr Valerius modestly observes that his main contribution to CF was “probably not this paper but the fact that in 1973 his sister-in-law, Annelise Hansen, was appointed nurse at the CF out patient clinic where she stayed for more than 30 years becoming almost an institution there”.

Annelise Hansen
Indeed I also remember Annelise very well and being very impressed by her when I visited the Copenhagen CF Centre for 2 days in 1997. Later, describing my visit to the outpatient clinic, I wrote “One is immediately conscious of a friendly and welcoming atmosphere and a good easy relationship between patients, parents and clinic staff. The senior clinic nurse, Annelise, has worked there for 28 years and combines the functions of clinic nurse manager, CF Nurse Specialist, and friend and adviser to the patients and their families” (A Visit to the Copenhagen CF Centre. Jim Littlewood. 1997. Pharmax Ltd). I recall asking Annelise if she had any problems dealing with both the children and the adults with CF. She replied that she and the staff had known the patients since they were small children and it was only natural that the relationship should continue when they became adults. Certainly the arrangement seemed to work very well in Copenhagen. However, in recent years an adult CF servcie has developed in Copenhagen.
In the present paper, Dr Valerius and the authors generously refer to our short letter to the Lancet in 1985 which was the first report on the use of inhaled colomycin to eradicate early Pseudomonas infection in CF and apparently stimulated their study. (Littlewood JM, Miller MG, Ghoneim AT, Ramsden CH. Nebulised colomycin for early Pseudomonas colonisation in cystic fibrosis. Lancet 1985; i: 865. [PubMed] Although only a letter, this letter was undoubtedly the most important publication of my career! It was good that the Copenhagen team confirmed our observations in their controlled trial. Prior to treating CF children with inhaled colomycin I had been impressed by a paper from the Sixties describing successful treatment of Pseudomonas aeruginosa respiratory infections in non-CF patients with a combination of IV and inhaled colistin (Halliday NP. Clinical Trials Journal. August 1967. 771-775) and also by an editorial by Wallace Herrell in Clinical Medicine (September 1968 18-19) of successful aerosolized treatment of 60 patients with gram negative pneumonia, 30 of whom were infected by P. aeruginosa, the majority of whom improved; P. aeruginosa was eliminated from their sputum. The editorial concluded – “Thus it would appear that sodium colistimethate can be administered safely as an aerosol and it appears to be quite effective in the treatment of pulmonary infections. including pneumonia, owing to sensitive organisms particulalry the Pseudomonas strains”. So after further discussions with the firm (Pharmax Ltd – now Forest Laboratories) as to the dose, nebuliser etc in 1983/4 we started to treat children as soon as P. aeruginosa was cultured from the monthly throat swabs. Even though the numbers in the present Valerius et al 1991 trial were small and the patients not formally randomised, this was certainly one of the most important papers of the decade and confirmed that early Pseudomonas infection could be eradicated with nebulised colomycin and oral ciprofloxacin.
It is difficult to understand why this trial from Copenhagen, which so clearly contradicted the previous widely held belief, that it was impossible to eradicate Pseudomonas once cultured, was not followed by the widespread introduction of early eradication treatment for P. aeruginosa. Fortunately a few centres in Europe did introduce early eradication treatment but they were a minority. The fact that early treatment of Pseudomonas was so slow to be introduced in the UK and much of Europe (with notable exceptions) and was still not recommended in the USA over a decade after this report, was difficult to explain.
It was predictable that these varied approaches to early treatment of Pseudomonas were reflected gradually in the markedly different prevalence of chronic
Fig 40. Christian Koch
Pseudomonas infection in different CF centres- this difference in the prevalence of chronic infection became increasingly obvious first in paediatric patients as time progressed (Frederiksen et al, 1996; Lee et al, 2003; Lebecque et al, 2006 – all below). It is of interest that even in 1998, reviewing the history of Pseudomonas infection in people with CF, a highly regarded US CF centre director wrote – “early administration with aerosol colistin may delay colonisation with P. aeruginosa. This intriguing observation has not been verified by prospective controlled studies” (Ramsey BW. Pediatrics 1998; Supplement: 210-213) – even though by this time early eradication of P. aeruginosa was widespread practice in Europe and already supported by many publications in addition to that of Valerius et al, 1991 from Copenhagen (Brett MM et al. Arch Dis Child 1992; 67:1086-1088.[PubMed]; Frederiksen B et al, Pediatr Pulmonol 1997; 23:330-335.[PubMed]; Weisemann HG, et al. Pediatr Pulmonol 1998; 25:88-92. [PubMed]; and later Munck A et al. Pediatr Pulmonol 2001; 32:288-292. [PubMed]
In 1997 I had the good fortune to interview the late Dr Christian Koch (figure 40), then the Medical Director of the Copenhagen CF centre, for a video. When I asked him at the end of the day what aspect of CF treatment he regarded as the most important, he thought for some time and then replied –“When I look back on what we’ve done all through the years that I’ve been involved with cystic fibrosis, I would say that the early treatment of Pseudomonas is probably the best thing that we have done for the patients. It becomes more and more clear that really what determines the long term course is whether you get Pseudomonas or not” .
1990 The use of ursodeoxycholic acid for CF related liver disease – the first treatment for CF related liver disease
1990 Colombo C, Setchell KD, Podda M, Crosignani A, Roda A, Curcio L, Ronchi M, Giunta A. Effects of ursodeoxycholic acid therapy for liver disease associated with cystic fibrosis. J Pediatr 1990; 117:482- 489. [PubMed]

Fig 41. Carla Colombo
The published report from Milan of data presented in 1989 by Carla Colombo (figure 41) at a memorable meeting of the European Working Group for CF in Prague reporting the favourable effect of ursodeoxycholic acid (URSO) treatment 10-15 mg /kg day in 9 patients with CF who had chronic liver disease (presumably the same patients as reported in Prague in 1989). Liver function tests improved significantly (AST- by 34%, alanine aminotransferase – by 41%, gamma gutamyl transpeptidase – by 41% and alkaline phosphatase– by 19%). Subsequently URSO was widely used in people with CF associated liver disease and represented one of the major advances in CF management for prior to URSO there was no treatment for CF liver disease. A further trail was published in 1996 (Colombo et al, 1996). Much information was published supporting URSO treatment at an early stage of liver involvement and eventually most people with CF with evidence of liver involvement were treated with URSO.
However, a Cochrane Review updated in 2008 concluded “There is insufficient evidence to justify its (URSO) routine use in cystic fibrosis”.In this writer’s opinion this conclusion was unfortunate as it could influence clinicians to withhold treatment from people with liver involvement. The following statement would have been more appropriate – “There is a considerable amount of evidence that URSO improves liver function in people with CF particularly if used at an early stage; but, as yet, there is no clinical trial that conforms to Cochrane standards to show this”.
1992 The first CF mice – essential for further CF research
1992 Snouweart JN, Brigman KK, Latour AM, Malouf NN, Boucher RC, Smithies O, et al. An animal model for cystic fibrosis made by gene targeting. Science 1992; 257:1083-1088. [PubMed] (August 1992)

Fig 42. Oliver Smithies
The authors generated a mouse line in which the cystic fibrosis transmembrane conductance regulator (CFTR) gene was mutated by gene targeting. Like patients with CF, mice lacking a functional CFTR gene (CFTR (-/-) mice) showed increased numbers of goblet cells and obstruction of glands with inspissated eosinophilic secretions. However, the most severe pathological changes in these mice were confined to the intestinal tract and gallbladder; there were only minor pathological alterations in the lungs and upper airways of the CFTR (-/-) mice. Possible explanations for the apparent lack of respiratory disease are the young age at which the animals were examined and the pathogen-free environment in which they were housed i.e. there had not been time to develop respiratory problems.The production of a “CF Mouse” was a major advance. Oliver Smithies was awarded a share of the Nobel Prize in 2007 for this work. The award was shared with Professor Martin Evans (Ratcliff et al, 1993 below). It is worth recalling that the lungs of CF infants who die in the newborn period also show little in the way of histological abnormality.
1992 Dorin JR, Dickinson P, Alton EWFW, Smith SN, Geddes DM, Stevenson BJ, et al. Cystic fibrosis in the mouse by targeted insertional mutagenesis. Nature 1992; 359:211-215. (Sept 1992). [PubMed]

Fig 43. Julia Dorin
To make this mouse the cystic fibrosis transmembrane conductance regulator gene was disrupted in embryonal stem cells using an insertional gene targeting vector. Germ-line chimaeras were derived and the offspring of heterozygous crosses studied. These homozygous mutant mice survived beyond weaning and in vivo electro physiology demonstrates the predicted defect in chloride ion transport and can distinguish between each genotype. Also histological analysis showed important hallmarks of human disease pathology, including abnormalities of the colon, lung and vas deferens.Dr Julia Dorin’s (figure 31) was one of the three groups to create a CF mouse model – the “Edinburgh mouse”. It provided a valid model system for the development and testing of therapies for cystic fibrosis patients and was a major advance. Dr Dorin is currently Programme Leader in Medical and Developmental Genetics at the MRC Human Genetics Unit in Edinburgh where her main research focus is on host defence peptides
1993 Ratcliff R, Evans MJ, Cuthbert AW, MacVinish LJ, Foster D, Anderson JR, et al. Production of a severe cystic fibrosis mutation in mice by gene targeting. Nature Genet 1993; 4:35-41. (May 1993). [PubMed]

Fig 44. Lesley McVinish
The authors used gene targeting in embryonic stem cells to introduce an HPRT mini-gene into the coding sequence of the murine cystic fibrosis gene (cftr). This insertion introduces a termination codon in frame with the cftr coding sequence to terminate prematurely the CFTR protein within the first nucleotide binding domain. Animals homozygous for the cftr disruption fail to thrive and display a range of symptoms including meconium ileus, distal intestinal obstructions, gastrointestinal mucus accumulation and blockage of pancreatic ducts. The animals also show lacrimal gland pathology. Tracheal and caecal transepithelial current measurements demonstrate the lack of a cAMP and can be used to study gene therapy strategies.This and the two preceding papers (Snouweart JN et al, 1992 above; Dorin et al, 1992 above) describing the creation of three types of CF mice were an essential development for further research into correction of the basic genetic defect.
Dr Lesley McVinish (figure 44) is Senior Teaching Associate, Dept of Pharmacology, Cambridge from 2004. For many years she

Fig 45. Sir Martin Evans
worked as a post doctoral researcher with Prof. Alan Cuthbert. Her research focussed on epithelial physiology and pharmacology, with a specific interest in cystic fibrosis. Subsequently she has held a number of important teaching and administrative posts in the University and also recieved the University’s Pilkiington Prize for Teaching in 2013.
Sir Martin Evans, (figure 45) first of Cambridge and then Cardiff, was awarded a share of the Nobel Prize in 2007 for his work on knockout mice – he shared the prize with Mario Capechi of Utah and Oliver Smithies of North Carolina. The Nobel citation describes the trio’s discovery for introducing specific gene modifications in mice by the use of embryonic stem cells. Martin Evans was the first to grow stem cells from early mouse embryos. He injected stem cells from one strain into the embryos of another strain then implanted the embryo into a surrogate mother leading to mosaic-celled mice
1993 Recognition of cross infection with potentially dangerous pathogens heralded a new era in CF care and had a profound influence on the treatment and social life of people with CF and their families
1993 Govan JR, Brown PH, Maddison J, Doherty CJ, Nelson JW, Dodd M, et al. Evidence for transmission of Pseudomonas cepacia by social contact in cystic fibrosis Lancet 1993; 342:15-19.[PubMed]

Fig 46. John Govan
Prof. John Govan of Edinburgh (figure 46) has for many years been a leading UK and international authority on CF microbiology. In this important paper he reported the definite transfer of B. cepacia infection between people with CF which finally convinced most CF clinicians. An analysis of isolates from 210 patients attending regional CF clinics in Edinburgh and Manchester between 1986 and 1992 showed that the main cause of increased isolations of P. cepacia from 1989 was the emergence of an epidemic strain that had spread between patients in both clinics. Epidemiological evidence indicated that social contact was important in spread of the epidemic strain within and between clinics. Guidelines to limit the acquisition of B. cepacia should not be restricted to patients in hospital, and that intimate or frequent social contact is associated with a high risk of cross-infection.Following this important paper the UK CF Trust advisory group of clinicians and microbiologists advised that strict segregation of B. cepacia-infected patients was essential as some clinics, even in 1993, were not yet segregating B. cepacia infected patients from others in the clinic.
Fortunately the widespread introduction of segregation from this time led to a steady reduction in the incidence of new B. cepacia infections and a reduction in the prevalence of chronic B. cepacia infections.Rosenstein & Hall reported pneumonia and septicemia due to Pseudomonas cepacia in a patient with CF (Rosenstein BJ, Hall DE. Johns Hopkins Med J 1980; 147:188-189) before the first report from Toronto describing major problems in treating the infection (Gold R et al. J Antimicrob Chemother 1983; 12 Suppl A: 331-336) and before the other major publication from Toronto (Isles A, et al. Pseudomonas cepacia infection in cystic fibrosis: an emerging problem. J Pediatr 1984; 104:206-210 below). This paper desribes how the prevalence of Pseudomonas cepacia infection in a population of approximately 500 patients with CF in the Toronto CF centre, increased from 10% in 1971 to 18% by 1981. The carriage of P. aeruginosa had remained unchanged at 70% to 80% over the same period. Patients infected with P. cepacia had greater impairment of pulmonary function than those with P. aeruginosa alone. A syndrome characterized by high fever, severe progressive respiratory failure, leukocytosis, and elevated erythrocyte sedimentation rate (“cepacia syndrome”) had occurred in eight patients over the previous three years, with a 62% fatality rate. Because P. cepacia strains are uniformly resistant to ticarcillin, piperacillin, and aminoglycosides, and because ceftazidime is ineffective despite in vitro activity, treatment of these infections was very difficult. Prevention of acquisition and effective treatment of P. cepacia in patients with cystic fibrosis had become a major problem in the Toronto clinic.The paper describes the devastating effect of the introduction of B. cepacia into a CF clinic population very clearly.In 1990 we published the first UK paper on B. cepacia in CF, having identified 11 patients with the infection over 6 years since 1984 but we found no evidence of obvious cross infection between our patients when we first identified the infection (Simmonds EJ, et al. Pseudomonas cepacia: a new pathogen in patients with cystic fibrosis referred to a large centre in the United Kingdom Arch Dis Child 1990; 65:874-877).The potential for spread between people with CF and the serious, often fatal, consequences (the so-called “cepacia syndrome”) were not generally appreciated in the UK until the early Nineties when John Govan’s 1993 publication documented definite person to person spread. This paper of John Govan’s had an immediate effect on the way B. cepacia patients were managed in the UK with regard to segregation from others with CF. From 1993 there was a general introduction of infection control measures in CF Centres in the UK; also there was an end to the North American CF holiday camps which were shown to be an important source of acquisition of the B. cepacia infection. In Leeds we had three patients who developed serious B. cepacia infections some months after visiting Canadian CF camps.
1994 Fibrosing colonopathy – a new and quite unexpected complication relating to the use of large doses of the recently introduced high strength pancreatic enzymes
1994 Smyth RL, van Velzen D, Smyth AR, Lloyd DA, Heaf DP. Strictures of the ascending colon in cystic fibrosis and high strength pancreatic enzymes. Lancet 1994; 343:85-86. [PubMed]

Fig 47. Rosiland Smyth
The first report of fibrosing colonopathy – a new, unexpected and serious complication subsequently shown to be related to the use of the new high strength pancreatic enzyme preparations that had been introduced during the previous 2 years (Pancrease HL, Creon 25,000, Nutrizym 22). The authors observed five children with CF, who presented over a period of two months, with meconium ileus equivalent that failed to respond to medical management. At surgery, four had a stricture in the ascending colon, and all had histopathological changes of post-ischemic ulceration repair, with mucosal and submucosal fibrosis. The only common change in the management of these children had been a switch from conventional enteric-coated pancreatic enzymes to a high-strength product between 12 and 15 months before presentation.
A subsequent case controlled study from these authors (Smyth et al, 1995 below) showed a significant correlation with both a high enzyme dose and the make of preparation – only those enzymes containing a covering of the copolymer eudragit being implicated. A similar study from the USA confirmed the association with high enzyme dosage but not with the copolymer covering (Friedman JP, FitzSimmons SC. Colonic strictures in patients with cystic fibrosis: results of a survey of 114 cystic fibrosis care centers in the United States. J Pediatr Gastroenterol Nutr 1996; 22:153-156. [PubMed] However, no further cases were reported or have been since reported, in patients taking the high strength Creon 25,000 preparation (which does not contain eudragit) even when given for 3 years in high doses to children (Connett et al, 1999 below).In the UK the recommendations of the Committee on Safety of Medicines to avoid doses of enzymes in excess of 10,000 IU lipase per kg day and preparations containing the copolymer eudragit in children, abolished the condition in the UK although a few cases continued to occur in the USA. The enzyme content of the various preparations are described in detail elsewhere (Littlewood JM, Wolfe SP, Conway SP. Diagnosis and treatment of malabsorption in cystic fibrosis. Pediatr Pulmonol 2006; 41:35-49.[PubMed]
1996 The first report of serious cross infection with P. aeruginosa in Liverpool proved by genomic finger printing techniques
1996 Cheng K, Smyth RL, Govan JRW, Doherty C, Winstanley C, Denning N, Heaf DP, Saene H van, Haret CA. Spread of ß-lactam resistant Pseudomonas aeruginosa in a cystic fibrosis clinic. Lancet 1996; 348:639-642. [PubMed]
A high proportion of children attending the Liverpool paediatric CF centre were found to be chronically infected with a P. aeruginosa that was resistant to ceftazidime and other beta-lactam antibiotics. Two genomic fingerprinting techniques were used to see if this had arisen from epidemic spread of a single strain. 92 (76.7%) of the 120 children attending the clinic were infected with P aeruginosa,and 65 (71%) of these 92 infected infants harboured isolates that were resistant to ceftazidime; 55 of the 65 children harboured the same epidemic strain – resistant to ceftazidime, azlocillin, and imipenem, and sensitive to tobramycin and ciprofloxacin. This study provides the first molecular evidence of a long-term “outbreak” of P. aeruginosa in a CF centre. The authors suggested that careful surveillance of the prevalence of antibiotic resistance in CF centres should be instituted with measures to prevent cross-infection. They suggested that anti-Pseudomonal monotherapy “should be considered with caution”. This lesson had already been learned in Copenhagen in the early Eighties (Pedersen et al, 1986 above) and in the past a number of writers had already cautioned against the use of monotherapy. Most CF centres at the time already followed the recommendation to use two antibiotics when treating exacerbations of Pseudomonas infection. However, prior to this outbreak, intravenous ceftazidime monotherapy had been routine in the Liverpool CF clinic. Although the use of two antibiotics is routine practice in most centres there is little published evidence to support the practice. It is cause for concern that, even after experience such as reported here, there is still discussion as to the use of one or two antibiotics for the treatment of respiratory exacerbations – even to the extent of a Cochrane review which failed to give firm advice to use two antibiotics!! (Ephick HE, Tan A. Single versus combination intravenous antibiotic therapy for people with cystic fibrosis. Cochrane Database of Systematic Reviews. 2005).This was a very important paper which highlighted the risk of cross infection with Pseudomonas aeruginosa in CF clinics, now clearly confirmed by genomic finger printing techniques. Later this highly transmissible “Liverpool epidemic strain” of Pseudomonas aeruginosa was reported elsewhere and subsequently spread to many other CF centres in the UK..
1997 The first suggestions that macrolides may have a favourable effect in people with CF who have P. aeruginosa infection
1997 Everard ML, Sly P, Brenan S, Ryan G. Macrolides antibiotics in diffuse panbronchiolitis and in cystic fibrosis. Eur Respir J 1997; 10:2926. [PubMed]

Fig 49. Adam Jaffe

Fig 48. Mark Everard
Mark Everard (figure 48) confirmed with me that this was the first report from Sheffield UK and Perth Western Australia showing the anti-inflammatory effect of macrolides in people with cystic fibrosis. Four of six patients with CF had significant reduction in IL-8 sputum levels after one month treatment with low dose (200 mg tds) oral erythromycin.Mark Everard believes the first report of the use of macrolides in CF was in a Japanese publication by Nakanishi et al in 1995 – “A 16-year-old boy was admitted to our hospital because of coughing, sputum, and exertional dyspnoea. Seven months after birth cystic fibrosis had been diagnosed. The chest roentgenogram on admission showed diffuse reticulonodular shadows and overinflation. Pulmonary function tests revealed obstructive and restrictive impairment. Erythromycin and Lomefloxacin were administered by mouth, and aminoglycosides were administered by inhalation. His symptoms were alleviated, and he is now an outpatient. In Japan, cystic fibrosis is rare, and this patient is extremely rare because he has grown up to be a 16-year-old. In this case, low-dose and long-term erythromycin administration was very effective”. (Nakanishi N, Ueda N, Kitade M, Moritaka T. A case of cystic fibrosis in a Japanese student. Jpn J Thoracic Dis 1995; 33:771-774.) [PubMed].
In a short report in 1998 Adam Jaffe (figure 49) and colleagues at the Royal Brompton showed an impressive improvement in seven severely affected children with CF. Daily azithromycin for more than 3 months increased FEV1 nad FVC by more than 11%
1998 Jaffe A, Francis J, Rosenthal M, Bush A. Long term azithromycin may improve lung function in children with cystic fibrosis. Lancet 1998; 351:420.
The beneficial effect of 600 mg/day of erythromycin for 1 to 12 months in chronic panbronchiolitis in Japanese patients had been reported previously (Nagai H et al. Respiration 1991; 58:145-9). [PubMed] The effect appeared to be independent of the presence of chronic Pseudomonas infection and an anti-inflammatory action was suggested (Fujii T et al. Thorax 1995; 50:1246-52; Hoiby N. Thorax 1994; 49:531-532; Kudoh S et al. Jpn J Thorac Dis 1987; 25:632-42; Kudoh S. et al. Am J Respir Crit Care Med 1998; 157:1829-32.; Kudoh et al, 1998.).These were important developments eventually leading to a number of large clinical trials and the widespread use of azithromycin in people with CF – undoubtedly one of the major clinical treatment advances of the Nineties and new Millennium (Equi et al, 2002 below; Saiman et al, 2003 both below). By 2005 nearly 54% of all the patients on the CF Foundation Registry were taking azithromycin.
1999 Benefits of intermittent inhaled tobramycin sufficient to achieve FDA approval and licensed in the USA
1999 Ramsey BW, Pepe MS, Quan JM, Otto KL, Montgomery AB, Williams-Warren J, Vasiljev-K M, Borowitz D, Bowman CM, Marshall BC, Marshall S, Smith AL. Intermittent administration of inhaled tobramycin in patients with cystic fibrosis. Cystic Fibrosis Inhaled Tobramycin Study Group. N Engl J Med 1999; 340:23-30. [PubMed]

Fig 50. Bonnie Ramsey
One of the most important clinical trials of the decade showing a significant benefit of inhaled preservative free tobramycin (TOBI) given during alternate four week cycles for 24 weeks to patients chronically infected with Pseudomonas aeruginosa. Treated patients had an average increase of FEV1 of 10% predicted at 20 weeks where as those on placebo had a 2% decline; also the treated patients had 23% fewer hospital admissions. The data was sufficient to convince the FDA and the drug was licensed.

Fig 51. Bruce Montgomery receiving a CFF award from Preston Campbel 111 for central role in the development of TOBI and other antibiotics
The introduction of TOBI, supported by this excellent clinical trial, was one of the major clinical advances of the decade and the culmination of work started in the late Eighties by Arnold Smith and others (Smith AL et al. 1989 above). By 2005 nearly 60% of people with CF in the US CFF Registry were receiving nebulised TOBI.
Although the cost (£10K per annum in the UK) has restricted the use of the TOBI preparation in the UK, inhaled anti-Pseudomonal antibiotics (colistin, tobramycin for injection and gentamicin) have been widely used in the UK for CF patients with chronic Pseudomonas infection since Margaret Hodson’s important 1981 paper (Hodson et al, 1981 above
Dr Bruce Montgomery (Figure 51) is now CEO of Cardeas Pharma, previously Senior Vice President and Head of Respiratory Therapeutics at Gilead Sciences, was closely involved with the development and trials of this preparation and has been involved subsequently with other new treatments including, more recently, nebulised aztreonam lysine for inhalation.
1996 Gentamicin and then PTC 124 (Ataluren) effective in reversing the effects of nonsense mutations. Unfortunately trials were equivocal and research is still progressing in 2019
1996 Howard M, Frizzell RA, Bedwell DM. Aminglycoside antibiotics restore CFTR function by overcoming premature stop mutationsNature Med 1996; 2:467-469. [PubMed
Howard and co-workers had demonstrated in cells carrying CFTR nonsense mutations, that gentamicin induced a dose-dependent increase in expression of full-length CFTR.
1997 Bedwell DM, Keanjak A, Bebok Z, Bubien JK, Tousson A, Clancy JP, Sorscher EJ. Suppression of a CFTR premature stop mutation in a bronchial epithelial cell line. Nature Med 1997; 3:1280-1284. [PubMed]

Fig 52. David Bedwell
Subsequently, Bedwell and co-workers showed in a CF bronchial epithelial cell line carrying the CFTR W1282X premature stop mutation, that gentamicin was capable of restoring CFTR expression on the apical membrane
Dr. Bedwell (figure 52), among a number of interests, is investigating whether pharmacological agents can be used to suppress nonsense mutations that cause genetic diseases. First, he is exploring whether this novel therapeutic approach can benefit patients with cystic fibrosis (CF) and Dr. Bedwell and his laboratory have published several papers demonstrating that drugs can suppress nonsense mutations in the CFTR gene in various CF experimental systems, including cultured CF cell lines and a CF mouse expressing a human CFTR-G542X transgene. He has constructed a new Cftr-G542X knock-in mouse model to explore this approach in a more physiologically relevant context.
2000 Wilschanski M, Famani C, Blau H, Rivlin J, Augarten A, Vital A, Kerem B, Kerem E. A pilot study of the effect of gentamicin on nasal potential difference measurements in cystic fibrosis patients carrying stop mutations. Am J Resp Crit Care 2000; 161:860-865. [PubMed]

Fig 53. Michael Wilschanski
This study, by Michael Wilschanski and colleagues from Haddash University Hospital, Israel, was the first to show that gentamicin in vivocould activate mutant CFTR in CF patients carrying stop mutations as had been suggested by Howard et al, (1996) and Bedwell et al, (1997).
Nine people with CF carrying stop mutations received gentamicin nasal drops for 14 days. The abnormal nasal potential difference improved after the gentamicin treatment suggesting that chloride transport had increased. The authors concluded that gentamicin may influence the underlying chloride transport abnormality in patients with CF carrying stop mutations (i.e. those mutations containing an X).
Aminoglycoside antibiotics can apparently increase the frequency of erroneous insertion of nonsense codons hence permitting the translation of CFTR alleles carrying missense mutations to continue reading to the end of the gene. It is appropriate that this study came from Israel as 64% of people with CF that country have at least one stop mutation. Intravenous gentamicin was also capable of producing small increases in CFTR conductance as judged by nasal PD measurements.Further work on these compounds in general Text or Topics – New Drugs
A NEW ERA OF CFTR MODULATORS – PLEASE ALSO SEE TOPIC SECTION
2006 VX-770 (Kalydeco/ivacaftor) – the first drug with significant potentiator effect on a specific CF mutation – G551D
2006 Van Goor F. Straley KS. Cao D. Gonzalez J. Hadida S. Hazlewood A. Joubran J. Knapp T. Makings LR. Miller M. Neuberger T. Olson E. Panchenko V. Rader J. Singh A. Stack JH. Tung R. Grootenhuis PD. Negulescu P.Rescue of DeltaF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. Am J Physiol_Lung C 2006; 290:L1117-30.[PubMed]

Fig 54. Frederick van Goor
Here, the authors describe two classes of novel, potent small molecules identified from screening compound libraries that restore the function of DeltaF508-CFTR in both recombinant cells and cultures of human bronchial epithelia isolated from CF patients.
The first class partially corrects the trafficking defect by facilitating exit from the endoplasmic reticulum and restores DeltaF508-CFTR-mediated Cl(-) transport to more than 10% of that observed in non-CF human bronchial epithelial cultures, a level expected to result in a clinical benefit in CF patients.
The second class of compounds potentiates cAMP-mediated gating of DeltaF508-CFTR and achieves single-channel activity similar to wild-type CFTR. The CFTR-activating effects of the two mechanisms are additive and support the rationale of a drug discovery strategy based on rescue of the basic genetic defect responsible for CF
Dr Fredeick van Goor (figure 40) is head of Biology CF Research Program, Vertex Pharmaceuticals. There are a number of accounts on the internet of the development of the Kalydeco (ivacaftor) by Vertex with considerable scientific, clinicial and financial support from the CF Foundation. The article by Matthew Herper in Forbes Magazine describes van Goor’s early involvement with the research.
2008 Accurso FJ, Rowe SM, Durie PR, Konstan MW, Dunitz J, Hornick DB, Sagel SD, Boyle MP, Uluer AZ, Upadhyay D, Ramsey BW, Freedman SD, Dong Q, Ahmed AM, Stone AJ, Olson ER, Ordenez CL, Clancy JP, Campbell PW, Ashlock MA. Interim results of Phase IIa study of VX-770 to evaluate safety, pharmokinetics and biomarkers of CFTR activity in cystic fibrosis subjects with G551D. Pediatr Pulmonol 2008; Suppl 31: 267 page 295. (Poster)

Fig 55. Frank Accurso
Results reported at the 2008 North American CF Conference of an interim analysis in 20 patients with the G551D mutation who received 14 days of oral VX-770, a potentiator of CFTR, 25mg, 75mg or 150mg 12 hourly or placebo. Those on highest dose of VX-770 (150mg twice daily) had an impressive response. They had increases in FEV1 of 10.1%, their sweat chloride fell from a mean of 95.5 to 53.2 mmol/l and their nasal potential difference showed significant changes towards normal.(-5.4mv in treated CF with -1.7mv in controls).This was an early but very encouraging result arising from the CF Foundation’s search for chemical activators of mutant CFTR. The change in sweat electrolytes is particularly impressive and this is the first drug to affect the level of sweat electrolytes in people with CF.A Phase II trial with 16 patients for 28 days started in 2008 and impressive progress was reported in 2009 (Boyle et al. Pediatr Pulmonol 2009; Suppl 32:287. Poster 217).A further Phase III trial was reported in November 2011 (below) which resulted in the VX-770 (now called Kalydeco) being approved by the FDA in January 2012.More details of these developments in Topics
Dr Frank Accurso (figure 55) Professor of Pediatrics and Head of Pulmonary Medicine in the University of Colorado. Following the selection of the University of Colorado as one of the Cystic Fibrosis Foundation’s Therapeutics Development Centers, Dr Accurso has been very active in the development and study of various new therapies and techniques for outcomes research
2011 Ramsey BW. Davies J. McElvaney NG. Tullis E. Bell SC. Drevinek P. Griese M. McKone EF. Wainwright CE. Konstan MW. Moss R. Ratjen F. Sermet-Gaudelus I. Rowe SM. Dong Q. Rodriguez S. Yen K. Ordonez C. Elborn JS. VX08-770-102 Study Group. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Eng J Med 2011; 365:1663-1672.[PubMed]
A randomized, double-blind, placebo-controlled trial to evaluate ivacaftor (VX-770), a CFTR potentiator, in subjects 12 years of age or older with cystic fibrosis and at least one G551D-CFTR mutation. Subjects were randomly assigned to receive 150 mg of ivacaftor every 12 hours (84 subjects, of whom 83 received at least one dose) or placebo (83, of whom 78 received at least one dose) for 48 weeks. The primary end point was the estimated mean change from baseline through week 24 in the percent of predicted forced expiratory volume in 1 second (FEV(1)).
RESULTS: The change from baseline through week 24 in the percent of predicted FEV(1) was greater by 10.6 percentage points in the ivacaftor group than in the placebo group (P<0.001). Effects on pulmonary function were noted by 2 weeks, and a significant treatment effect was maintained through week 48. Subjects receiving ivacaftor were 55% less likely to have a pulmonary exacerbation than were patients receiving placebo, through week 48 (P<0.001). In addition, through week 48, subjects in the ivacaftor group scored 8.6 points higher than did subjects in the placebo group on the respiratory-symptoms domain of the Cystic Fibrosis Questionnaire-revised instrument (a 100-point scale, with higher numbers indicating a lower effect of symptoms on the patient’s quality of life) (P<0.001). By 48 weeks, patients treated with ivacaftor had gained, on average, 2.7 kg more weight than had patients receiving placebo (P<0.001). The change from baseline through week 48 in the concentration of sweat chloride, a measure of CFTR activity, with ivacaftor as compared with placebo was -48.1 mmol per liter (P<0.001). The incidence of adverse events was similar with ivacaftor and placebo, with a lower proportion of serious adverse events with ivacaftor than with placebo (24% vs. 42%).
The authors concluded that ivacaftor was associated with improvements in lung function at 2 weeks that were sustained through 48 weeks. Substantial improvements were also observed in the risk of pulmonary exacerbations, patient-reported respiratory symptoms, weight, and concentration of sweat chloride. A quite remarkable result.
VX-809 (LUMACAFTOR) – a new CFTR corrector
2011 Van Goor F. Hadida S. Grootenhuis PD. Burton B. Stack JH. Straley KS. Decker CJ. Miller M. McCartney J. Olson ER. Wine JJ. Frizzell RA. Ashlock M. Negulescu PA. Correction of the F508del-CFTR protein processing defect in vitro by investigational drug VX-809. Proc Natl Acad Sci USA 2011; 108:18843-8.[PubMed] Most patients with CF carry the F508del CFTR mutation, which causes defective CFTR protein folding and processing in the endoplasmic reticulum, resulting in minimal amounts of CFTR at the cell surface. One strategy to treat these patients is to correct the processing of F508del-CFTR with small molecules. Here the authors describe the in vitro pharmacology of VX-809, a CFTR corrector that was advanced into clinical development for the treatment of CF. In cultured human bronchial epithelial cells isolated from patients with CF homozygous for F508del, VX-809 improved F508del-CFTR processing in the endoplasmic reticulum and enhanced chloride secretion to approximately 14% of non-CF human bronchial epithelial cells (EC(50), 81 +/- 19 nM), a level associated with mild CF in patients with less disruptive CFTR mutations. F508del-CFTR corrected by VX-809 exhibited biochemical and functional characteristics similar to normal CFTR, including biochemical susceptibility to proteolysis, residence time in the plasma membrane, and single-channel open probability. VX-809 was more efficacious and selective for CFTR than previously reported CFTR correctors. VX-809 represents a class of CFTR corrector that specifically addresses the underlying processing defect in F508del-CFTR.
2012 Clancy JP. Rowe SM. Accurso FJ. Aitken ML. Amin RS. Ashlock MA. Ballmann M. Boyle MP. Bronsveld I. Campbell PW. De Boeck K. Donaldson SH. Dorkin HL. Dunitz JM. Durie PR. Jain M. Leonard A. McCoy KS. Moss RB. Pilewski JM. Rosenbluth DB. Rubenstein RC. Schechter MS. Botfield M. Ordonez CL. Spencer-Green GT. Vernillet L. Wisseh S. Yen K. Konstan MW. Results of a Phase IIa study of VX-809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for the F508Del-CFTR mutation. Thorax 2012; 67:12-18. [PubMed]
VX-809,(LUMCAFTOR) a cystic fibrosis transmembrane conductance regulator (CFTR) modulator, has been shown to increase the cell surface density of functional F508del-CFTR in vitro. This is a randomised, double-blind, placebo-controlled study evaluated the safety, tolerability and pharmacodynamics of VX-809 in adult patients with cystic fibrosis (n=89) who were homozygous for the F508del-CFTR mutation. Subjects were randomised to one of four VX-809 28 day dose groups (25, 50, 100 and 200 mg) or matching placebo.
The type and incidence of adverse events were similar among VX-809- and placebo-treated subjects. Respiratory events were the most commonly reported and led to discontinuation by one subject in each active treatment arm. Pharmacokinetic data supported a once-daily oral dosing regimen. Pharmacodynamic data suggested that VX-809 improved CFTR function in at least one organ (sweat gland). VX-809 reduced elevated sweat chloride values in a dose-dependent manner (p=0.0013) that was statistically significant in the 100 and 200 mg dose groups.There was no statistically significant improvement in CFTR function in the nasal epithelium as measured by nasal potential difference, nor were there statistically significant changes in lung function or patient-reported outcomes. No maturation of immature F508del-CFTR was detected in the subgroup that provided rectal biopsy specimens.
The authors concluded that VX-809 (LUMACAFTOR) had a similar adverse event profile to placebo for 28 days in F508del-CFTR homozygous patients, and demonstrated biological activity with positive impact on CFTR function in the sweat gland. Additional data were needed to determine how improvements detected in CFTR function secondary to VX-809 in the sweat gland relate to those measurable in the respiratory tract and to long-term measures of clinical benefi
Ivacaftor effective in children aged 6-11years
2013 Davies JC, Wainwright CE, Canny GJ, Chilvers MA, Howenstine MS, Munck A, Mainz JG, Rodriguez S, Li H, Yen K, Ordonez CL, Ahrens R. VX08-770-103 (ENVISION) Study Group. Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. Am J Resp Crit Care 2013; 187:1219-25. [PubMed]

Jane Davies
Children received oral ivacaftor 150 mg (n = 26) or placebo (n = 26) every 12 hours for 48 weeks. Despite near-normal mean baseline values in FEV1, patients receiving ivacaftor had a significant increase in percent predicted FEV1 from baseline through Week 24 versus placebo group (treatment effect, 12.5 percentage points; P < 0.001). Effects on pulmonary function were evident by 2 weeks, and a significant treatment effect was maintained through Week 48. Patients treated with ivacaftor gained, on average, 2.8 kg more than those receiving placebo at Week 48 (P < 0.001). The change from baseline through Week 48 in the concentration of sweat chloride, a measure of CFTR activity, with ivacaftor was -53.5 mmol/L (P < 0.001) versus placebo. The incidence of adverse events was similar in the two groups.
These quite dramatic results further confirm the very significant benefit from ivacaftor even in those children with a G551D mutation who have normal or near normal respiratory function. The authors concluded that in patients who are younger and healthier than those in previously studied populations, ivacaftor demonstrated a significant improvement in pulmonary function, weight, and CFTR activity compared with placebo.
– These trials reporting the effects of ivacaftor in adults and here in children must surely be the most sensational and important publications that have appeared to date relating to the treatment of CF.
First press report of effectiveness of triple therapy – VX 445 (elexaacaftor), VX 809 (lumacaftor) and VX 770 (ivacaftor)
2014 Adam Feuerstein. Vertex Pharma Cystic Fibrosis Combo Therapy Hits Key Endpoints in Two Pivotal Trials. A report in The Street on 24.6.14
Combination therapy of two Vertex Pharmaceuticals (VRTX 445) drugs, VX-809 (lumacaftor) and Kalydeco (ivacaftor VX-770), produced a statistically significant improvement in lung function for patients with the most common form of cystic fibrosis (F508del-CFTR). The two phase III studies dubbed “TRAFFIC” and “TRANSPORT” both met their primary endpoints separately. When the studies were pooled together, cystic fibrosis patients treated with the higher of two doses of VX-809 (also known as Lumacaftor) and Kalydeco saw their lung function improve by 3.3% on an absolute basis compared to placebo.
Vertex’s combination regimen also achieved statistical significance versus placebo on two key secondary endpoints of the phase III studies — a 30% and 39% reduction in pulmonary exacerbations and improvements in weight gain (body mass index.) [Vertex shared the study results with the writer of this abstract on June 23rd under embargo.]
First major trial of lumacaftor (VX 809) and ivacaftor (VX 770) – ORKAMBI
2014 Boyle MP. Bell SC. Konstan MW. McColley SA. Rowe SM. Rietschel E. Huang X. Waltz D. Patel NR. Rodman D. VX09-809-102 study group. A CFTR corrector (lumacaftor) and a CFTR potentiator (ivacaftor) for treatment of patients with cystic fibrosis who have a phe508del CFTR mutation: a phase 2 randomised controlled trial. Lancet Respir Med 2014; 2(7):527-38. [PubMed]

Michael Boyle
This important multicentre study tested combination treatment with lumacaftor, an investigational CFTR corrector that increases trafficking of phe508del CFTR to the cell surface, and ivacaftor, a CFTR potentiator that enhances chloride transport of CFTR on the cell surface.
The authors assessed three successive cohorts, with the results of each cohort informing dose selection for the subsequent cohort. They recruited patients from 24 cystic fibrosis centres in Australia, Belgium, Germany, New Zealand, and the USA. Eligibility criteria were: confirmed diagnosis of cystic fibrosis, age at least 18 years, and a forced expiratory volume in 1s (FEV1) of 40% or more than predicted.
Cohort 1 included phe508del CFTR homozygous patients randomly assigned to either lumacaftor 200 mg once per day for 14 days followed by addition of ivacaftor 150 mg or 250 mg every 12 h for 7 days, or 21 days of placebo.
Together, cohorts 2 and 3 included phe508del CFTR homozygous and heterozygous patients, randomly assigned to either 56 days of lumacaftor (cohort 2: 200 mg, 400 mg, or 600 mg once per day, cohort 3: 400 mg every 12 h) with ivacaftor 250 mg every 12 h added after 28 days, or 56 days of placebo.
The primary outcomes for all cohorts were change in sweat chloride concentration during the combination treatment period in the intention-to-treat population and safety (laboratory measurements and adverse events). (The study is registered with ClinicalTrials.gov, number NCT01225211, and EudraCT, number 2010-020413-90).
Cohort 1 included 64 participants. Cohort 2 and 3 combined contained 96 phe508del CFTR homozygous patients and 28 compound heterozygotes. Treatment with lumacaftor 200 mg once daily and ivacaftor 250 mg every 12 h decreased mean sweat chloride concentration by 9.1 mmol/L (p<0.001) during the combination treatment period in cohort 1.
In cohorts 2 and 3, mean sweat chloride concentration did not decrease significantly during combination treatment in any group. Frequency and nature of adverse events were much the same in the treatment and placebo groups during the combination treatment period; the most commonly reported events were respiratory. 12 of 97 participants had chest tightness or dyspnoea during treatment with lumacaftor alone. In pre-planned secondary analyses, a significant decrease in sweat chloride concentration occurred in the treatment groups between day 1 and day 56 (lumacaftor 400 mg once per day group -9.1 mmol/L, p<0.001; lumacaftor 600 mg once per day group -8.9 mmol/L, p<0.001; lumacaftor 400 mg every 12 h group -10.3 mmol/L, p=0.002). These changes were significantly greater than the change in the placebo group. In cohort 2, the lumacaftor 600 mg once per day significantly improved FEV1 from day 1 to 56 (difference compared with placebo group: +5.6 percentage points, p=0.013), primarily during the combination period. In cohort 3, FEV1 did not change significantly across the entire study period compared with placebo (difference +4.2 percentage points, p=0.132), but did during the combination period (difference +7.7 percentage points, p=0003). Phe508del CFTR heterozygous patients did not have a significant improvement in FEV1.
This trial provided evidence that combination lumacaftor and ivacaftor (Orkambi) improves FEV1 for patients with cystic fibrosis who are homozygous for phe508del CFTR, with a modest effect on sweat chloride concentration. These results support the further exploration of combination lumacaftor and ivacaftor as a treatment in this setting. The abstract of this important study and rather complex study is reproduced in full.
2015 Wainwright CE, Elborn JS, Ramsey BW, Marigowda G, Huang X, Cipolli M, Colombo C, Davies JC, De Boeck K, Flume PA, Konstan MW, McColley SA, McCoy K, McKone EF, Munck A, Ratjen F, Rowe SM, Waltz D, Boyle MP; TRAFFIC Study Group; TRANSPORT Study Group. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N Engl J Med. 2015 Jul 16;373(3):220-31. doi: 10.1056/NEJMoa1409547. Epub 2015 May 17. [PubMed]

Claire Wainwright
Two phase 3, randomised, double-blind, placebo-controlled studies were designed to assess the effects of lumacaftor (VX-809), a CFTR corrector, in combination with ivacaftor (VX-770), a CFTR potentiator, (Orkambi) in patients 12 years of age or older who had cystic fibrosis and were homozygous for the Phe508del CFTR mutation. In both studies, patients were randomly assigned to receive either lumacaftor (600 mg once daily or 400 mg every 12 hours) in combination with ivacaftor (250 mg every 12 hours) or matched placebo for 24 weeks. The primary end point was the absolute change from baseline in the percentage of predicted forced expiratory volume in 1 second (FEV1) at week 24.
A total of 1108 patients underwent randomization and received study drug. The mean baseline FEV1 was 61% of the predicted value. In both studies, there were significant improvements in the primary end point in both lumacaftor-ivacaftor dose groups; the difference between active treatment and placebo with respect to the mean absolute improvement in the percentage of predicted FEV1 ranged from 2.6 to 4.0 percentage points (P<0.001), which corresponded to a mean relative treatment difference of 4.3 to 6.7% (P<0.001). Pooled analyses showed that the rate of pulmonary exacerbations was 30 to 39% lower in the lumacaftor-ivacaftor groups than in the placebo group; the rate of events leading to hospitalization or the use of intravenous antibiotics was lower in the lumacaftor-ivacaftor groups as well. The incidence of adverse events was generally similar in the lumacaftor-ivacaftor and placebo groups. The rate of discontinuation due to an adverse event was 4.2% among patients who received lumacaftor-ivacaftor versus 1.6% among those who received placebo.
These data show that lumacaftor in combination with ivacaftor provided a benefit for patients with cystic fibrosis homozygous for the Phe508del CFTR mutation.; benefit was a modest 3% improvement in FEV1 and 35% decrease in pulmonary exacerbations
Ivacaftor (VX 770) effective in children aged 2 – 5 years with a CFTR gating mutation
2016 Davies JC; Cunningham S; Harris WT; Lapey A; Regelmann WE; Sawicki GS; Southern KW; Robertson S; Green Y; Cooke J; Rosenfeld M; KIWI Study Group. Safety, pharmacokinetics, and pharmacodynamics of ivacaftor in patients aged 2-5 years with cystic fibrosis and a CFTR gating mutation (KIWI): an open-label, single-arm study.The Lancet Respir Med 2016; 4(2):107-15.[PubMed]
Jane Davies
Ivacaftor has been shown to be a safe, effective treatment for cystic fibrosis in patients aged 6 years or older with a CFTR gating mutation. The authors aimed to assess the safety, pharmacokinetics, and pharmacodynamics of ivacaftor in children aged 2-5 years.In the two-part KIWI study, we enrolled children aged 2-5 years weighing 8 kg or more with a confirmed diagnosis of cystic fibrosis and a CFTR gating mutation on at least one allele from 15 hospitals in the USA, UK, and Canada. Participants received oral ivacaftor 50 mg (if bodyweight <14 kg) or 75 mg (if bodyweight >14 kg) every 12 h for 4 days in part A (to establish the short-term safety of doses for subsequent assessment in part B), and then for 24 weeks in part B (to assess safety and longer-term pharmacodynamics). Children could participate in both or just one part of the study. Primary outcomes were pharmacokinetics and safety, analysed in all patients who received at least one dose of ivacaftor. Secondary outcomes were absolute change from baseline in sweat chloride concentrations and bodyweight, body-mass index (BMI), and height Z scores, and pharmacokinetic parameter estimation of ivacaftor. This study is registered with ClinicalTrials.gov, number NCT01705145.
Between Jan 8, 2013, and March 1, 2013, nine patients were enrolled onto part A of the study, all of whom completed the 4 day treatment period, and eight of whom took part in part B. Between June 28, 2013, and Sept 26, 2013, 34 patients were enrolled in part B, 33 of whom completed the 24 week treatment period. All patients received at least one dose of ivacaftor. Results of ivacaftor pharmacokinetics suggested that exposure was similar to that reported in adults (median Cmin were 536 ng/mL for the 50 mg dose; 580 ng/mL for the 75 mg dose; median ivacaftor AUC values were 9840 ng x h/mL and 10 200 ng x h/mL, respectively). Common adverse events in part B included cough (in 19 [56%] of 34 patients) and vomiting (in ten [29%]). Five (15%) patients had liver function test (LFT) results that were more than eight times higher than the upper limit of normal, four of whom had study drug interrupted, and one of whom had study drug discontinued. Six (18%) of 34 patients had seven serious adverse events; a raised concentration of transaminases was the only serious adverse event regarded as related to ivacaftor and the only adverse event that resulted in study treatment discontinuation. At week 24, in patients for whom we had data, sweat chloride had changed from baseline by a mean of -46.9 mmol/L (SD 26.2, p<0.0001), weight Z score by 0.2 (0.3; p<0.0001), BMI Z score by 0.4 (0.4, p<0.0001), and height Z score by -0.01 (0.3; p=0.84).
Ivacaftor at doses of 50 mg and 75 mg seems to be safe in children aged 2-5 years with cystic fibrosis with a gating mutation followed up for 24 weeks, although the frequency of elevated LFTs suggests that monitoring should be frequent in young children, particularly those with a history of elevated LFTs. Results of an ongoing extension study assessing durability of these effects and longer-term safety are warranted.
– This study confirmed that ivacaftor was safe and effective for children aged 2-5 years. Full abstract included in view of importance.
See comment from Yammine S et al, 2016 below for comment on the dose and effect on liver function in young children taking Ivacaftor.
Lumacaftor (VX 809) and ivacaftor (VX 770) – ORKAMBI effective in children aged 6 – 11 years
2016 Milla CE, Ratjen F, Marigowda G, Liu F, Waltz D, Rosenfeld M; VX13-809-011 Part B Investigator Group. Lumacaftor/Ivacaftor in Patients Aged 6-11 Years With Cystic Fibrosis Homozygous for F508del-CFTR. Am J Respir Crit Care Med. 2016 Nov 2. [Epub ahead of print] [PubMed]
Combination lumacaftor/ivacaftor (Orkambi) as been shown to improve lung function and other endpoints in patients aged ≥12 years with cystic fibrosis homozygous for F508del-CFTR but has not been assessed in younger patients.
This open-label phase 3 trial evaluated the safety, tolerability, pharmacodynamics, and efficacy of lumacaftor/ivacaftor combination therapy in patients aged 6-11 years with cystic fibrosis homozygous for F508del-CFTR. Patients (N = 58) received 200 mg lumacaftor/250 mg ivacaftor orally every 12 hours for 24 weeks in addition to their existing cystic fibrosis medications.
Lumacaftor/ivacaftor was well tolerated; the safety profile was generally similar to that observed in larger lumacaftor/ivacaftor trials in older patients. Four patients discontinued (two due to drug-related adverse events: elevated liver transaminases, n=1; rash, n=1). No safety concerns were associated with spirometry. No significant changes in % predicted FEV1 were observed (change from baseline at Week 24, +2.5 percentage points; 95% CI, -0.2 to 5.2; P = 0.0671). At Week 24, significant improvements from baseline were observed in sweat chloride (-24.8 mmol/L; 95% CI, -29.1 to -20.5; P < 0.0001), BMI z score (+0.15; 95% CI, 0.08 to 0.22; P < 0.0001), Cystic Fibrosis Questionnaire-Revised respiratory domain score (+5.4; 95% CI, 1.4 to 9.4; P = 0.0085), and lung clearance index 2.5 (-0.88; 95% CI, -1.40 to -0.37; P = 0.0018).
– Lumacaftor/ivacaftor was well tolerated in this young population; no new safety concerns were identified. Improvements in lung clearance index, sweat chloride, nutritional status, and health-related quality of life were observed after 24 weeks of treatment.
2016 Rowe SM, McColley SA, Rietschel E, Li X, Bell SC, Konstan MW, Marigowda G, Waltz D, Boyle MP; VX09‐809‐102 Study Group.Lumacaftor/Ivacaftor Treatment of Patients with Cystic Fibrosis Heterozygous for F508del-CFTR. Ann Am Thorac Soc. 2016 Nov 29. [Epub ahead of print] [PubMed]
Lumacaftor/ivacaftor (Orkambi) treatment (≤28 days) in patients with cystic fibrosis (CF) heterozygous for F508del-CFTR did not improve lung function. This study was to evaluate an optimised lumacaftor/ivacaftor dosing regimen with longer duration in a cohort of patients heterozygous for F508del-CFTR.
Patients age ≥18 years with a confirmed CF diagnosis and percent predicted forced expiratory volume in 1 second (ppFEV1) of 40 to 90 were randomised to lumacaftor/ivacaftor (400 mg/250 mg every 12 hours) or placebo daily for 56 days. Primary outcomes were change in ppFEV1 at day 56 and safety. Other disease markers were evaluated.Of 126 patients; 119 (94.4%) completed the study. Lumacaftor/ivacaftor was well tolerated, although chest tightness and dyspnea occurred more frequently with active treatment than with placebo (27.4% vs 14.3% and 14.5% vs 6.3%, respectively). Mean (SD) ppFEV1 at baseline was 62.9 (14.3) [active treatment] and 60.1 (14.0) [placebo]. Absolute change in ppFEV1 (least squares mean [SE]) at day 56 was -0.6 (0.8) [active treatment] and -1.2 (0.8) percentage points [placebo] (P = 0.60). CF respiratory symptom scores improved by a mean of 5.7 points versus a decrease of -0.8 for placebo (P < 0.01). No changes in body mass index (BMI) occurred. Change from baseline in sweat chloride (least squares mean [SE]) at day 56 was -11.8 (1.3) mmol/L [active treatment] and -0.8 (1.2) mmol/L [placebo] (P < 0.0001).
The authors concluded sweat chloride and respiratory symptom scores improved with lumacaftor/ivacaftor, though no meaningful benefit was seen in ppFEV1 and BMI in patients heterozygous for F508del-CFTR.
– This is a modest effect for patients heterozygous for F508del which would be unlikely to result in the treatment being recommended for patients heterozygous for F508del – certainly in the UK where, at this time, the combination therapy was not even approved by NICE for patients homozygous for F508del.
Tezacaftor (VX-661)/ivacaftor (VX-770) = (SYMDEKO) active for homozygous and heterozygous for F508del
2017 Donaldson SH, Pilewski JM, Griese M, Cooke J, Viswanathan L, Tullis E, Davies JappearingC, Lekstrom-Himes JA, Wang LT; VX11-661-101 Study Group. Tezacaftor/Ivacaftor in Subjects with Cystic Fibrosis and F508del/F508del-CFTR or F508del/G551D-CFTR. Am J Respir Crit Care Med. 2017 Sep 20. doi: 10.1164/rccm.201704-0717OC. [Epub ahead of print] [Pubmed]

Scott H Donaldson
Tezacaftor (formerly VX-661) is an investigational small molecule that improves processing and trafficking of the cystic fibrosis transmembrane conductance regulator (CFTR) in vitro, and improves CFTR function alone and in combination with ivacaftor. This is a study to evaluate safety and efficacy of tezacaftor monotherapy and tezacaftor/ivacaftor combination therapy in subjects with CF homozygous for F508del or compound heterozygous for F508del and G551D. This was a randomized, placebo-controlled, double-blind, multicenter, phase 2 study (NCT01531673). Subjects homozygous for F508del received tezacaftor (10 mg to 150 mg) qday alone or in combination with ivacaftor 150 mg q12h in a dose escalation phase, as well as in a dosage regimen-testing phase. Subjects compound heterozygous for F508del and G551D taking physician prescribed ivacaftor received tezacaftor 100 mg qday.
Primary endpoints were safety through day 56 and change in sweat chloride from baseline through day 28. Secondary endpoints included change in percent predicted FEV1 (ppFEV1) from baseline through day 28 and pharmacokinetics. The incidence of adverse events was similar across treatment arms.
Tezacaftor 100 mg qday/ivacaftor 150 mg q12h resulted in a 6.04 mmol/L decrease in sweat chloride and 3.75 percentage point increase in ppFEV¬1 in subjects homozygous for F508del and a 7.02 mmol/L decrease in sweat chloride and 4.60 percentage point increase in ppFEV¬1 in subjects compound heterozygous for F508del and G551D from baseline through day 28 (P < 0.05 for all).
The authors consider these results support continued clinical development of tezacaftor 100 mg qday in combination with ivacaftor 150 mg q12h in subjects with CF.
Dr. Scott H Donaldson is Associate Professor of Medicine, Pulmonary Diseases and Critical Care Medicine, University of North Carolina School of Medicine.
Lumacaftor/ivacaftor (ORKAMBI) was safe in and tolerated by children aged 6 – 11 years of age
2017 Milla CE, Ratjen F, Marigowda G, Liu F, Waltz D, Rosenfeld M; VX13-809-011 Part B Investigator Group. Lumacaftor/Ivacaftor in Patients Aged 6-11 Years with Cystic Fibrosis and Homozygous for F508del-CFTR. Am J Respir Crit Care Med. 2017 Apr 1;195(7):912-920. doi: 10.1164/rccm.201608-1754OC [Pubmed]
Combination lumacaftor/ivacaftor has been shown to improve lung function and other endpoints in patients aged 12 years and older with cystic fibrosis and homozygous for F508del-CFTR, but it has not been assessed in younger patients. In this open-label phase III trial, we evaluated the safety, tolerability, pharmacodynamics, and efficacy of lumacaftor/ivacaftor combination therapy in patients aged 6-11 years with cystic fibrosis who were homozygous for F508del-CFTR. Patients (N = 58) received 200 mg lumacaftor/250 mg ivacaftor orally every 12 hours for 24 weeks in addition to their existing cystic fibrosis medications.
Lumacaftor/ivacaftor was well tolerated; the safety profile was generally similar to that observed in larger lumacaftor/ivacaftor trials with older patients. Four patients discontinued (two because of drug-related adverse events: elevated liver transaminases, n = 1; rash, n = 1). No safety concerns were associated with spirometry. No significant changes in percent predicted FEV1 were observed (change from baseline at Week 24, +2.5 percentage points; 95% confidence interval [CI], -0.2 to 5.2; P = 0.0671). At Week 24, significant improvements from baseline were observed in sweat chloride (-24.8 mmol/L; 95% CI, -29.1 to -20.5; P < 0.0001), body mass index z score (+0.15; 95% CI, 0.08 to 0.22; P < 0.0001), Cystic Fibrosis Questionnaire-Revised respiratory domain score (+5.4; 95% CI, 1.4 to 9.4; P = 0.0085), and lung clearance index based on lung volume turnover required to reach 2.5% of starting N2concentration (-0.88; 95% CI, -1.40 to -0.37; P = 0.0018).
The authors concluded Lumacaftor/ivacaftor was well tolerated in this young population; no new safety concerns were identified. Improvements in lung clearance index, sweat chloride, nutritional status, and health-related quality of life were observed after 24 weeks of treatment. There was no significant effect on the FEV1
2017 Ratjen F, Hug C, Marigowda G, Tian S, Huang X, Stanojevic S, Milla CE, Robinson PD, Waltz D, Davies JC; VX14-809-109 investigator group. Efficacy and safety of lumacaftor and ivacaftor in patients aged 6-11 years with cystic fibrosis homozygous for F508del-CFTR: a randomised, placebo-controlled phase 3 trial. Lancet Respir Med. 2017 Jul;5(7):557-567. doi: 10.1016/S2213-2600(17)30215-1. Epub 2017 Jun 9. [Pubmed]

Felix Ratjen
Lumacaftor and ivacaftor combination treatment showed efficacy in patients aged 12 years or older with cystic fibrosis homozygous for F508del-cystic fibrosis transmembrane conductance regulator (CFTR) in placebo-controlled studies and for patients aged 6-11 years with cystic fibrosis homozygous for F508del-CFTR in an open-label study. The authors report efficacy and safety of lumacaftor and ivacaftor in patients with cystic fibrosis aged 6-11 years homozygous for F508del-CFTR.
In this phase 3, randomised, double-blind, placebo-controlled, multicentre study, patients were enrolled at 54 hospitals and medical centres in nine countries (the USA, Australia, Belgium, Canada, Denmark, France, Germany, Sweden, and the UK). Eligible patients weighed at least 15 kg, with a confirmed diagnosis of cystic fibrosis, per cent predicted forced expiratory volume in 1 s (FEV1) of 70 or more, and lung clearance index 2·5 (LCI 2·5) of 7·5 or more at screening (values less than these thresholds were permitted at day 1). All patients were tested for CFTR genotype at screening; eligible patients had to have the F508del-CFTR mutation on both alleles. Exclusion criteria included any comorbidity or laboratory abnormality that might confound the study results or pose additional risk to the patient. Patients were stratified by weight (<25 kg vs. ≥25 kg) and ppFEV1 severity (<90 vs. ≥90) determined at the screening visit, and randomly assigned 1:1 to treatment using an interactive web response system to receive 200 mg lumacaftor and 250 mg ivacaftor every 12 hours or placebo for 24 weeks. The primary endpoint was the mean absolute change in LCI 2·5 from all on-treatment study visits up to and including week 24. All randomly assigned patients who were exposed to any amount of study drug, with treatment assignment as assigned were included in primary and other efficacy analyses. All patients who were exposed to any amount of study drug, with treatment assignment as treated, were included in the safety analysis. This study was registered with ClinicalTrials.gov, number NCT02514473.
Between July 23, 2015, and Sept 20, 2016, a total of 206 patients were enrolled and randomly assigned to receive lumacaftor and ivacaftor (n=104) or placebo (n=102). Two randomly assigned patients were never dosed with study drug (one in the placebo arm due to ineligibility arising from a streptococcal throat infection and one in the lumacaftor and ivacaftor arm due to withdrawal based on refusal to provide blood tests) and were not included in the analyses.
103 patients received at least one dose of lumacaftor and ivacaftor and 101 patients received at least one dose of placebo. For the primary endpoint, the average absolute change in LCI2·5 from baseline over all study visits up to and including the week 24 visit, least squares mean difference was -1·09 units (95% CI -1·43 to -0·75, p<0·0001) for lumacaftor and ivacaftor versus placebo. For the key secondary endpoint of sweat chloride concentration, the least squares mean difference versus placebo was -20·8 mmol/L (95% CI -23·4 to -18·2, average absolute change at day 15/week 4; p<0·0001). The least squares mean difference compared with placebo in absolute change in ppFEV1 from all on-treatment study visits until week 24 was 2·4 (95% CI 0·4-4·4, p=0·0182). 196 (96%) of 204 patients reported adverse events, most of which were mild (87 [43%]) or moderate (98 [48%]). Treatment was discontinued due to adverse events in three (3%) of 103 patients in the lumacaftor and ivacaftor group and two (2%) of 101 patients in the placebo group. Serious adverse events were reported in 13 (13%) of 103 patients in the lumacaftor and ivacaftor group and 11 (11%) of 101 patients in the placebo group.
The authors concluded that treatment with lumacaftor and ivacaftor was associated with statistically significant improvements in lung function, as measured by LCI2·5 and ppFEV1, versus placebo in patients aged 6-11 years with cystic fibrosis homozygous for F508del-CFTR. The overall safety profile was consistent with previous phase 3 studies of lumacaftor and ivacaftor.
– The first important major study on lumacaftor/ivacaftor children aged 6 to 11 years. Again with modest effect on respiratory function.
2017 Rowe SM, McColley SA, Rietschel E, Li X, Bell SC, Konstan MW, Marigowda G, Waltz D, Boyle MP; VX09-809-102 Study Group. Lumacaftor/Ivacaftor Treatment of Patients with Cystic Fibrosis Heterozygous for F508del-CFTR. Ann Am Thorac Soc. 2017 Feb;14(2):213-219. doi: 10.1513/Annals ATS.201609-689OC.[Pubmed]

Steven Rowe
In a prior study, lumacaftor/ivacaftor treatment (≤28 d) (Orkambi) in patients with cystic fibrosis (CF) heterozygous for F508del-CFTR did not improve lung function. This present study was to evaluate an optimized lumacaftor/ivacaftor dosing regimen with a longer duration in a cohort of patients heterozygous for F508del-CFTR. Lumacaftor/ivacaftor (400 mg/250 mg every 12 h) or placebo daily for 56 days.
Sweat chloride and respiratory symptom scores improved with lumacaftor/ivacaftor, though no meaningful benefit was seen in ppFEV1 or body mass index in patients heterozygous for F508del-CFTR even with optimised dosing.
Tezacaftor (Vx 661)/ivacaftor (VX 770) (SYMDEKO) had moderate but definite effect on sweat chloride and respiratory function
2018 Donaldson SH, Pilewski JM, Griese M, Cooke J, Viswanathan L, Tullis E, Davies JC, Lekstrom-Himes JA, Wang LT5; VX11-661-101 Study Group. Tezacaftor/Ivacaftor in Subjects with Cystic Fibrosis and F508del/F508del-CFTR or F508del/G551D-CFTR.Am J Respir Crit Care Med. 2018 Jan 15;197(2):214-224. doi: 10.1164/rccm.201704-0717OC.[Pubmed]
Scott H Donaldson
A study to evaluate the safety and efficacy of tezacaftor (formerly VX-661) monotherapy and of tezacaftor/ivacaftor combination therapy in subjects with cystic fibrosis homozygous for F508del or compound heterozygous for F508del and G551D. This was a randomized, placebo-controlled, double-blind, multicenter, phase 2 study. Subjects homozygous for F508del received tezacaftor (10 to 150 mg) every day alone or in combination with ivacaftor (150 mg every 12 h) in a dose escalation phase, as well as in a dosage regimen testing phase. Subjects compound heterozygous for F508del and G551D, taking physician-prescribed ivacaftor, received tezacaftor (100 mg every day).
Primary endpoints were safety through Day 56 and change in sweat chloride from baseline through Day 28. Secondary endpoints included change in per cent predicted FEV1 (ppFEV1) from baseline through Day 28 and pharmacokinetics. The incidence of adverse events was similar across treatment arms. Tezacaftor (100 mg every day)/ivacaftor (150 mg every 12 h) resulted in a 6.04 mmol/L decrease in sweat chloride and 3.75 percentage point increase in ppFEV1 in subjects homozygous for F508del, and a 7.02 mmol/L decrease in sweat chloride and 4.60 percentage point increase in ppFEV1 in subjects compound heterozygous for F508del and G551D from baseline through Day 28 (P < 0.05 for all).
The authors considered these results support continued clinical development of tezacaftor (100 mg every day) in combination with ivacaftor (150 mg every 12 h) in subjects with cystic fibrosis.
Dr. Steven Rowe (figure) is a pulmonologist in Birmingham, Alabama and is affiliated with University of Alabama at Birmingham Hospital
Ivacaftor (VX 770) shown to be safe and effective in chidden aged 12- 24 months
2018 Rosenfeld M,Wainwright CE,Higgins M,Wang LT,McKee C,Campbell D,Tian S,Schneider J,Cunningham S,Davies JC;ARRIVAL study group.Ivacaftor treatment ofcystic fibrosisin children aged 12 to <24 months and with a CFTR gating mutation (ARRIVAL): a phase 3 single-arm study. Lancet Respir Med.2018Jun 6. pii: S2213-2600(18)30202-9. doi: 10.1016/S2213-2600(18)30202-9. [Epub ahead of print] [Pubmed]

Margaret Rosenfield
The ARRIVAL study is a phase 3, single-arm, two-part, multicentre study. Eligible children were aged 12 to <24 months at enrolment and had a confirmed diagnosis of cystic fibrosis and a CFTR gating mutation on at least one allele and could participate in one or both parts of the study.
Children received 50 mg (bodyweight 7 to <14 kg) or 75 mg (bodyweight ≥14 to <25 kg) ivacaftor orally every 12 h. In study part A, children received ivacaftor for 3 days plus one morning. In study part B, children received 24 weeks of treatment. Seven children were enrolled in part A, of whom five received 50 mg and two received 75 mg ivacaftor. All completed treatment. Of 19 children enrolled in part B, including one from part A, all received 50 mg ivacaftor and 18 completed treatment (one withdrew because of difficulty with blood draws). All children received at least one dose of ivacaftor. Pharmacokinetics indicated exposure was similar to that in children aged 2 to <6 years and adults. No children discontinued because of adverse events or safety findings. In part A, three (43%) of seven children had treatment-emergent adverse events, all of which were mild and deemed not to be or unlikely to be related to ivacaftor. By 24 weeks in part B, treatment-emergent adverse events had been reported in 18 (95%) of 19 children, of which most were mild or moderate and the most frequent was cough (14 [74%] children). Two children in part B had four serious adverse events: one had constipation (possibly related to ivacaftor), distal intestinal obstruction syndrome, and eczema herpeticum, and one had persistent cough, all needing hospital admission. In five (28%) of 18 children aspartate or alanine aminotransferase concentrations rose to more than three times the upper limit of normal (to more than eight times in two children with concurrent infections). At week 24, the mean absolute change from baseline in sweat chloride concentration was -73·5 (SD 17·5) mmol/L. Growth parameters for age were normal at baseline and at week 24. At week 24, concentrations of faecal elastase-1 had increased and concentrations of immunoreactive trypsinogen had decreased from baseline. Mean serum lipase and amylase were raised at baseline and rapidly decreased after treatment was started
The authors concluded that ivacaftor was generally safe and well tolerated in children aged 12 to <24 months for up to 24 weeks and was associated with rapid and sustained reductions in sweat chloride concentrations. Improvements in biomarkers of pancreatic function suggest that ivacaftor preserves exocrine pancreatic function if started early. The study is continuing in infants younger than 12 months.
Lumacaftor/ivacaftor (Orkambi) is safe and well tolerated by children aged 12 to <24 months
McNamara JJ, McColley SA, Marigowda G, Liu F3 Tian S, Owen CA, Stiles D, Li C, Waltz D, Wang LT, Sawicki GS. Safety, pharmacokinetics, and pharmacodynamics of lumacaftor and ivacaftor combination therapy in children aged 2-5 years with cystic fibrosis homozygous for F508del-CFTR: an open-label phase 3 study.Lancet Respir Med. 2019 Jan 24. pii: S2213-2600(18)30460-0. doi: 10.1016/S2213-2600(18)30460-0. [Epub ahead of print] [Pubmed]
-

John MacNamara
- The efficacy, safety, and tolerability of lumacaftor and ivacaftor are established in patients aged 6 years and older with cystic fibrosis, homozygous for the F508del-CFTR mutation. We assessed the safety, pharmacokinetics, pharmacodynamics, and efficacy of lumacaftor and ivacaftor in children aged 2-5 years.
In this multicentre, phase 3, open-label, two-part study, we enrolled children aged 2-5 years, weighing at least 8 kg at enrolment, with a confirmed diagnosis of cystic fibrosis who were homozygous for the F508del-CFTR mutation. Children received lumacaftor 100 mg and ivacaftor 125 mg (bodyweight <14 kg) or lumacaftor 150 mg and ivacaftor 188 mg (bodyweight ≥14 kg) orally every 12 h for 15 days in part A (to assess pharmacokinetics and safety) and for 24 weeks in part B (to assess safety, pharmacokinetics, pharmacodynamics, and efficacy). Children could participate in part A, part B, or both. Children were enrolled into part A at five sites in the USA and into part B at 20 sites in North America (USA, 17 sites; Canada, three sites). The primary endpoints of the study were the pharmacokinetics (part A) and safety (part B) of lumacaftor and ivacaftor; all analyses were done in children who received at least one dose of lumacaftor and ivacaftor. Secondary endpoints in part A were safety and pharmacokinetics of the metabolites of lumacaftor and ivacaftor, and in part B included pharmacokinetics in children who received at least one dose of lumacaftor and ivacaftor and absolute changes from baseline in sweat chloride concentration, growth parameters, and markers of pancreatic function. This study is registered with ClinicalTrials.gov, number NCT02797132.
The study was done from May 13, 2016, to Sept 8, 2017. 12 children enrolled in part A, 11 of whom completed the 15-day treatment period and enrolled in part B. 60 children enrolled in part B, 56 of whom completed the 24-week treatment period. Safety and pharmacokinetics were consistent with the well characterised safety profile of lumacaftor and ivacaftor. In part B, most children (59 [98%] of 60 children) had one or more treatment-emergent adverse events; most events were mild to moderate in severity. The most common adverse events were cough (38 [63%] of 60), vomiting (17 [28%]), pyrexia (17 [28%]), and rhinorrhoea (15 [25%]). Serious adverse events occurred in four children: infective pulmonary exacerbation of cystic fibrosis (n=2), gastroenteritis viral (n=1), and constipation (n=1). Three (5%) of 60 children discontinued treatment because of elevated serum aminotransferase concentrations. Mean sweat chloride concentrations decreased by 31·7 mmol/L, biomarkers of pancreatic function improved (fecal elastase-1 concentrations increased and serum immunoreactive trypsinogen concentrations decreased), and growth parameters increased at week 24.
Lumacaftor and ivacaftor were generally safe and well tolerated in children aged 2-5 years with cystic fibrosis for 24 weeks. Efficacy findings also suggest that early intervention with lumacaftor and ivacaftor has the potential to modify the course of the disease.
THE NEW TRIPLE THERAPY – A MAJOR STEP FORWARD. Elexacaftor (VX 445) / Tezacaftor (VX 661)/Ivacaftor (VX 770) = (TRIKAFTA in USA – KAFTRIO in Europe) has a significant effect in patients both DF508 homozygous and heterozygous
2018 Keating D, Marigowda G, Burr L, Daines C, Mall MA, McKone EF, Ramsey BW, Rowe SM, Sass LA, Tullis E, McKee CM, Moskowitz SM, Robertson S, Savage J, Simard C, Van Goor F, Waltz D, Xuan F, Young T, Taylor-Cousar JL; VX16-445-001 Study Group. VX-445-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles N Engl J Med. 2018 Oct 25;379(17):1612-1620. doi: 10.1056/NEJMoa1807120. Epub 2018 Oct 18. [Pubmed]

Dominic Keating
VX-445 is a next-generation cystic fibrosis transmembrane conductance regulator (CFTR) corrector designed to restore Phe508del CFTR protein function in patients with cystic fibrosis when administered with tezacaftor and ivacaftor (VX-445-tezacaftor-ivacaftor). The authors evaluated the effects of VX-445-tezacaftor-ivacaftor on Phe508del CFTR protein processing, trafficking, and chloride transport in human bronchial epithelial cells. On the basis of in vitro activity, a randomized, placebo-controlled, double-blind, dose-ranging, phase 2 trial was conducted to evaluate oral VX-445-tezacaftor-ivacaftor in patients heterozygous for the Phe508del CFTR mutation and a minimal-function mutation (Phe508del-MF) and in patients homozygous for the Phe508del CFTR mutation (Phe508del-Phe508del) after tezacaftor-ivacaftor run-in. Primary end points were safety and absolute change in percentage of predicted forced expiratory volume in 1 second (FEV1) from baseline.
Results. In vitro, VX-445-tezacaftor-ivacaftor significantly improved Phe508del CFTR protein processing, trafficking, and chloride transport to a greater extent than any two of these agents in dual combination. In patients with cystic fibrosis, VX-445-tezacaftor-ivacaftor had an acceptable safety and side-effect profile. Most adverse events were mild or moderate. The treatment also resulted in an increased percentage of predicted FEV1 of up to 13.8 points in the Phe508del-MF group (P<0.001). In patients in the Phe508del-Phe508del group, who were already receiving tezacaftor-ivacaftor, the addition of VX-445 resulted in an 11.0-point increase in the percentage of predicted FEV1 (P<0.001). In both groups, there was a decrease in sweat chloride concentrations and improvement in the respiratory domain score on the Cystic Fibrosis Questionnaire-Revised.
The authors concluded the use of VX-445-tezacaftor-ivacaftor to target Phe508del CFTR protein resulted in increased CFTR function in vitro and translated to improvements in patients with cystic fibrosis with one or two Phe508del alleles. This approach has the potential to treat the underlying cause of cystic fibrosis in approximately 90% of patients. (Clinical trials NCT03227471 ; and EudraCT number, 2017-000797-11
Dominic T Keating is Respiratory Physician at the Alfred Hospital · Department of Allergy, Immunology & Respiratory Medicine (AIRmed) Australia, Melbourne
Heijerman HGM, McKone EF, Downey DG, Van Braeckel E, Rowe SM, Tullis E Mall MA, Welter JJ, Ramsey BW, McKee CM, Marigowda G, Moskowitz SM, Waltz D, Sosnay PR, Simard C, Ahluwalia N, Xuan F, Zhang Y, Taylor-Cousar JL, McCoy KS; VX17-445-103 Trial Group. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: a double-blind, randomised, phase 3 trial. Collaborators (45). Lancet. 2019 Oct 30. pii: S0140-6736(19)32597-8. doi: 10.1016/S0140-6736(19)32597-8. [Epub ahead of print] Full copy available [Pubmed]

Harry Heijerman
Cystic fibrosis transmembrane conductance regulator (CFTR) modulators correct the basic defect caused by CFTR mutations. Improvements in health outcomes have been achieved with the combination of a CFTR corrector and potentiator in people with cystic fibrosis homozygous for the F508del mutation. The addition of elexacaftor (VX-445), a next-generation CFTR corrector, to tezacaftor plus ivacaftor further improved F508del-CFTR function and clinical outcomes in a phase 2 study in people with cystic fibrosis homozygous for the F508del mutation.
This phase 3, multicentre, randomised, double-blind, active-controlled trial of elexacaftor in combination with tezacaftor plus ivacaftor was done at 44 sites in four countries. Eligible participants were those with cystic fibrosis homozygous for the F508del mutation, aged 12 years or older with stable disease, and with a percentage predicted forced expiratory volume in 1 s (ppFEV1) of 40-90%, inclusive. After a 4-week tezacaftor plus ivacaftor run-in period, participants were randomly assigned (1:1) to 4 weeks of elexacaftor 200 mg orally once daily plus tezacaftor 100 mg orally once daily plus ivacaftor 150 mg orally every 12 h versus tezacaftor 100 mg orally once daily plus ivacaftor 150 mg orally every 12 h alone. The primary outcome was the absolute change from baseline (measured at the end of the tezacaftor plus ivacaftor run-in) in ppFEV1 at week 4. Key secondary outcomes were absolute change in sweat chloride and Cystic Fibrosis Questionnaire-Revised respiratory domain (CFQ-R RD) score. This study is registered with ClinicalTrials.gov, NCT03525548.
Between Aug 3 and Dec 28, 2018, 113 participants were enrolled. Following the run-in, 107 participants were randomly assigned (55 in the elexacaftor plus tezacaftor plus ivacaftor group and 52 in the tezacaftor plus ivacaftor group) and completed the 4-week treatment period. The elexacaftor plus tezacaftor plus ivacaftor group had improvements in the primary outcome of ppFEV1 (least squares mean [LSM] treatment difference of 10·0 percentage points [95% CI 7·4 to 12·6], p<0·0001) and the key secondary outcomes of sweat chloride concentration (LSM treatment difference -45·1 mmol/L [95% CI -50·1 to -40·1], p<0·0001), and CFQ-R RD score (LSM treatment difference 17·4 points [95% CI 11·8 to 23·0], p<0·0001) compared with the tezacaftor plus ivacaftor group. The triple combination regimen was well tolerated, with no discontinuations. Most adverse events were mild or moderate; serious adverse events occurred in two (4%) participants receiving elexacaftor plus tezacaftor plus ivacaftor and in one (2%) receiving tezacaftor plus ivacaftor.
The authors concluded Elexacaftor plus tezacaftor plus ivacaftor provided clinically robust benefit compared with tezacaftor plus ivacaftor alone, with a favourable safety profile, and shows the potential to lead to transformative improvements in the lives of people with cystic fibrosis who are homozygous for the F508del mutation.
Dr Harry Heijerman is at the Department of Pulmonology, University Medical Center Utrecht, Utrecht, Netherlands.
Middleton PG, Mall MA, Drevinek P, Lands LC, McKone EF, Polineni D, Ramsay BW, Taylor0Cousar JL, Tullis E, Vermeulen F, marifwada G, Mosowski SM, Nair N, Savage J, Simard C, Tian S, Waltz D, Xuan F, Jain Rassha for the VX17-445-i102 Study Group. Elexacaftor-tezacaftor-iavacaftor for cystic fibrosis with a single The 508del allele. N Eng J Med 2019; 381:1809-19. [Pubmed]

Peter Middleton
Cystic fibrosis is caused by mutations in the gene encoding the cystic fibrosis transmembrane conductance regulator (CFTR) protein, and nearly 90% of patients have at least one copy of the Phe508del CFTR mutation. In a phase 2 trial involving patients who were heterozygous for the Phe508del CFTR mutation and a minimal- function mutation (Phe508del–minimal function genotype), the next-generation CFTR corrector elexacaftor, in combination with tezacaftor and ivacaftor, improved Phe508del CFTR function and clinical outcomes.
the authors conducted a phase 3, randomised, double-blind, placebo-controlled trial to confirm the efficacy and safety of elexacaftor–tezacaftor–ivacaftor in patients 12 years of age or older with cystic fibrosis with Phe508del–minimal function geno- types. Patients were randomly assigned to receive elexacaftor–tezacaftor–ivacaftor or placebo for 24 weeks. The primary end point was absolute change from baseline in percentage of predicted forced expiratory volume in 1 second (FEV1) at week 4.
A total of 403 patients underwent randomisation and received at least one dose of active treatment or placebo. Elexacaftor–tezacaftor–ivacaftor, relative to placebo, resulted in a percentage of predicted FEV1 that was 13.8 points higher at 4 weeks and 14.3 points higher through 24 weeks, a rate of pulmonary exacerbations that was 63% lower, a respiratory domain score on the Cystic Fibrosis Questionnaire– Revised (range, 0 to 100, with higher scores indicating a higher patient-reported quality of life with regard to respiratory symptoms; minimum clinically important difference, 4 points) that was 20.2 points higher, and a sweat chloride concentration that was 41.8 mmol per litre lower (P<0.001 for all comparisons). Elexacaftor– tezacaftor–ivacaftor was generally safe and had an acceptable side-effect profile. Most patients had adverse events that were mild or moderate. Adverse events leading to discontinuation of the trial regimen occurred in 1% of the patients in the elexacaftor–tezacaftor–ivacaftor group.
Conclusion – Elexacaftor–tezacaftor–ivacaftor was efficacious in patients with cystic fibrosis with Phe508del–minimal function genotypes, in whom previous CFTR modulator regimens were ineffective. (VX17-445-102 ClinicalTrials.gov number, NCT03525444.)
Professor Peter Middleton is Clinical Professor of Respiratory and Sleep Medicine Westmead Hospital and CF Research Group, Ludwig Engel Centre for Respiratory Research, University of Sydney.
Review and meta-analysis of CFTR corrector and potentiator trials up to the present
Wu HX, Zhu M, Xiong XF, Wei J, Zhuo KQ, Cheng DY. Efficacy and Safety of CFTR Corrector and Potentiator Combination Therapy in Patients with Cystic Fibrosis for the F508del-CFTR Homozygous Mutation: A Systematic Review and Meta-analysis. Adv Ther. 2019 Feb;36(2):451-461. doi: 10.1007/s12325-018-0860-4. Epub 2018 Dec 15. [Pubmed]
The authors performed a systematic review and meta-analysis of randomized controlled trials (RCTs) to evaluate the efficacy and safety of combination therapy on lung function, nutritional status, clinical score and safety in CF for the F508del-CFTR homozygous mutation. Five RCTs, including a total of 1637 participants with the F508del-CFTR homozygous mutation who accepted CFTR corrector and potentiator combination therapy along with basic treatment were enrolled in this analysis. Primary analysis revealed that combination therapy improved the percent of predicted FEV1 (ppFEV1) (MD 2.38, 1.62-3.15, P < 0.00001), Cystic Fibrosis Questionnaire-Revised (CFQ-R) respiratory domain score (MD 2.59, 0.96-4.22, P = 0.002) and body-mass index (BMI) (MD 0.21, 0.03-0.39, P = 0.02). In the secondary analysis, combination therapy had no impact on the number of participants reporting adverse events (OR 0.88, 0.58-1.33, P = 0.53), but increased the proportion of discontinued treatments due to adverse events (OR 2.71, 1.3-5.63, P = 0.008).
The authors concluded potentiator combination therapy effectively improves lung function, nutritional status and clinical score in CF patients with the F508del-CFTR homozygous mutation, and has an acceptable safety profile.
From the Department of Respiratory and Critical Care Medicine, West China Hospital, Sichuan University, Chengdu, China.