The Forties


Forties
“Fibrocystic disease of the pancreas” gradually recognised as a relatively common, generalised, inherited and fatal disorder
SUMMARY OF THE DECADE
During the Forties few doctors anywhere had even heard of cystic fibrosis. During the first 5 years of the decade Europe was at war; many countries were involved or even under occupation. So it was not surprising that little attention was given to the recently described “Fibrocystic Disease of the Pancreas”. Undoubtedly, the Second World War, the demands of other diseases and the lack of awareness of CF and the lack of medical care and poor health of a large number of children in the population accounted for the paucity of publications on CF from the UK and mainland Europe during the decade. On the positive side, as a result of the need for penicillin to treat the wounded during the war, its development was implemented and its use as an antibiotic introduced. Prior to the availability of antibiotics few infants with CF survived infancy and early childhood.

Fig.1. Aneurin Bevan
In the UK, during the Forties after the war, there were major changes in society and attitudes – not least in the provision of free medical care in the UK through the National Health Service introduced in 1948 under the guidance of Aneurin Bevan, (Fig.1) the then Minister of Health. Undoubtedly the one good thing to come out of the war in the UK was the provision of free health care for all.

Fig. 2 Archie Norman
Dr Archie Norman (Fig. 2) of the Hospital for Sick Children, Great Ormond Street, London later recalled – “for those of us who had come back from the war (the late Forties) it was an extremely exciting period. So many new advances in medicine; paediatrics itself had become accepted as a real branch of medicine, and not a junior part; there was the discovery of antibiotics that totally changed our attitude to infectious disease which was of particular importance, of course, to cystic fibrosis; last, but not least, was the advent of the National Health Service in 1948 – we could prescribe drugs without worrying whether the family could afford them, a matter of immense importance in a persistent long-term disease such as cystic fibrosis” (Norman AP, Wellcome Witness Seminar, London, 2002 below). This last facility was to prove of immense benefit for people with cystic fibrosis as more survived for longer and treatments became increasingly more expensive.

Fig. 3. Sydney Farber
During the decade Dr Sydney Farber (Fig. 3), Chief of Pathology at the Children’s Hospital, Boston, recognised CF as being a generalised disorder affecting organs other than the pancreas and he is attributed with introducing the term “mucoviscidosis”. Farber accurately summarized the secondary consequences of the CF defect as – “the respiratory tract damage therefore depends on primary obstruction by thick mucus, failure of proper lubrication of ciliated epithelium and secondary Staphylococcal infection” (Farber, 1943 below). Farber was a senior colleague of the legendary Harry Shwachman in Boston, who, with Paul di Sant’Agnese and Dorothy Andersen of New York, were the leading authorities on CF in the USA at that time.
There were various suggestions as to the aetiology of CF such as a maternal illness during pregnancy with some toxic effects on the fetus. In 1944 Howard, studying two families with an unusual number of deaths, suggested the possibility of a heterozygous inheritance (Howard, 1944. below) and in 1946 Dorothy Andersen and her paediatric colleague in New York, Dr Hodges, investigating 113 families, were among the first of a number of authors to conclude, correctly, that there was a Mendelian recessive mode of inheritance but commented that – “the disease, although hereditary, requires more than one factor for expression” (Andersen & Hodges, 1946 below); others agreed, including the geneticist Dr Cedric Carter of London, and his colleagues Archie Norman and Martin Bodian (Bodian, 1952 below).
The radical change in the Forties, for a few children in the USA recognized as having CF, was the introduction of penicillin in 1944 and later other antibiotics, aureomycin (1948) and terramycin (1950), without which these children did not survive. Although sulphonamides (so-called “M&B” after the pharmaceutical firm May and Baker) became available from the mid-Thirties, there were variable opinions concerning its value in cystic fibrosis. There were no effective antibiotics for Staphylococcus aureus, the main respiratory pathogen in CF at the time, until a limited amount of penicillin became available in the mid-Forties. In 1943, working in New York, Paul di Sant’Agnese had obtained a small quantity of penicillin from the US army and treated a few children with CF with “dramatic” results, whether the antibiotic was given by aerosol or by intramuscular injection (a very painful treatment I can personally remember!). At that time the most common pathogen was a penicillin-sensitive Staphylococcus aureus (di Sant’Agnese et al, 1946 below).

FIg. 4a Dr Jerry Kelleher

Prof. Monty Losowsky
The treatment in the Forties, in the few places where there was any, focused mainly on nutrition and there was still a widespread belief that malnutrition, and in particular vitamin A deficiency with its effect on the structure of the respiratory epithelium (causing squamous metaplasia), in some way contributed to the expression of the basic defect causing the intractable respiratory infections. There was much discussion as to whether the changes in other organs were part of a primary disease process or secondary to various nutritional deficiencies.
It did not seem unreasonable to believe, as did Dorothy Andersen, that damage to the respiratory epithelium from vitamin A deficiency would predispose the airways to progressive infection which, due to these changes in the respiratory epithelium, was impossible to eradicate. I confess this possibility seemed perfectly reasonable to me even in the Seventies and, in part, influenced the choice of our first CF research project in Leeds in the late Seventies on the vitamin status of our few children with cystic fibrosis (Congden et al, 1981 below). The other factor which influenced our choice was access to the considerable nutritional and research expertise of our colleagues Professor Monty Losowsky (Fig. 4) and Dr Jerry Kelleher (Fig.4a) in the University Department of Medicine at St James’s University Hospital, Leeds. This was a fortunate choice as it was the start of many years of fruitful collaboration between our CF unit and Professor Losowsky’s Department of Medicine at St James’s University Hospital, Leeds. (Described in detail elsewhere in the History section of this website “The History of the CF Services in Leeds. The Origins and Development of the Leeds Regional Cystic Fibrosis Unit Pt. 1 & Pt. 2)

Dorothy Andersen
Dorothy Andersen (Fig. 5), although a pathologist, wrote widely on treatment and advised – “a low fat, high protein diet with a liberal allowance of vegetables, fruits and sugar and moderate restriction of starch. Supplementary vitamin A is essential and pancreatin and vitamin B complex are given” (Andersen, 1945 below).
In 1946, di Sant’Agnese also stressed the importance of diet and attributed the improved prognosis to “an appropriate diet begun promptly and continued consistently, use of sulphadiazine during the stage of chronic cough and the use of nebulised penicillin” – interestingly he later came to the view that “sulfa drugs were totally ineffective”. It is also interesting that in 1984 Shwachman recalled that “before 1948 there was no antibiotic for CF. There was penicillin and streptomycin: they were not effective. So 1948 was the year of aureomycin and in 1949 we had terramycin” (Fanos JH, 2008 below). The need for early treatment, before significant lung damage, represents the subsequent view of many experienced CF clinicians.
During the Forties there appeared reports of small series of children with this recently recognised condition. These were usually derived from a retrospective review of the numerous post-mortems on children performed during previous years at some of the larger children’s hospitals in North America. These reviews were undoubtedly prompted by Andersen’s 1938 paper rather than any of the previous publications and all the cases were identified by their having typical pancreatic changes of CF noted at autopsy.
So in the Forties CF was still a new disease and unknown to most people. Milton Graub, a paediatrician and parent who later became president of the US CF Foundation, recalls that even in 1950, doctors were slow to diagnose his two year old son as having cystic fibrosis. Eventually, he called Dr Andersen on a Sunday morning; she was in her laboratory and saw the boy the next day and diagnosed his cystic fibrosis.
Papers published in the Forties
1941 Beazell JM, Schmidt CR, Ivy AC. The diagnosis and treatment of achylia pancreatica. J Am Med Assoc 1941; 116:2735-2739.
This classic paper, from Professor Ivy’s group, was said to initiate the modern treatment of pancreatic insufficiency. At the start, the authors state “Since the number of well studied patients are few and since oral pancreatic enzyme therapy in man is not generally considered to be of value, a view that is inconsistent with most of the results with animal experiments, we have undertaken to determine the effectiveness of oral pancreatic enzyme therapy in human patients with achylia pancreatica”.
Four patients with non-CF pancreatic insufficiency were studied in detail with estimation of faecal fat and nitrogen and also estimation of their duodenal enzymes – which were totally absent. Enzyme replacement therapy resulted in an average reduction in daily faecal nitrogen excretion of 62% (7.84 to 2.99 g per day) and lipids of 63.3% (74.4 to 27.3 g per day). All the patients gained weight (6 to 40 lbs) over 2.5 to 36 months. The authors note that Dorothy Andersen had already shown that pancreatic enzyme therapy improved the intestinal malabsorption of children with cystic fibrosis (Andersen DH. J Pediatr 1939; 15:763).

Fig. 6. A C Ivy. From The Paradox of the Pancreas. With permission of Professor Irvin Modlin
Professor AC Ivy (1893-1978) (Fig. 6) was a famous physiologist who, with Oldberg, discovered cholecystokinin in 1928 and apparently published some 2000 scientific articles.
– There was considerable discussion for many years among paediatricians as to whether pancreatic replacement therapy was worth using – some paediatricians considered that the unpleasant taste of the crude pancreatic extract did more harm than good in putting children off their food to achieve only modest improvements in absorption. However, all agreed that the introduction of the acid resistant microspheres (Pancrease and Creon) in the early Eighties represented a major advance in treatment.
1941 Jeffrey FW. Cystic fibrosis of the pancreas. Canad Med Assoc J 1941; 45:224-229. Full text available. PubMed
“Cystic fibrosis of the pancreas is a definite disease entity with a rather typical symptomatology” – so begins this excellent review from Ottawa of the present knowledge of CF with a review of 88 patients from the literature and a report of two further examples. The typical pancreatic changes were described for it was on the basis of these changes in pancreatic histology that the condition was differentiated from other forms of coeliac syndrome. The author classified the cases as described by Andersen into those dying in the neonatal period, those less than and those more than 6 months – all in the latter two groups succumbed to respiratory infection.
1942 Farber S. Experimental production of achylia pancreatica: summary. Am J Dis Child 1942; 64:953-954.
Children subsequently shown to have pancreatic lesions were classified into three groups. Those who died at a few weeks, usually from meconium ileus; those who died in the first year with nutritional disease often obscured by respiratory disease and those with primarily coeliac signs who later died of respiratory disease (also Farber, 1943 below).
1942 Andersen DH. Pancreatic enzymes in duodenal juice in celiac syndrome. Am J Dis Child 1942; 63:643.
The first report that estimation of duodenal trypsin was a reliable diagnostic test for CF as there was a wide difference between the concentration of trypsin in the duodenum in CF and in other gastrointestinal conditions and controls. Andersen noted that in normal infants trypsin and lipase were present from birth and amylase from 2 or 3 months.
– Trypsin was more reliable than lipase although levels of the enzymes may be reduced in marasmus or severe dehydration; this was also noted by Shwachman. This finding was confirmed many years later as a temporary phenomenon in gluten-induced coeliac disease, when it was attributed to lack of substrate to form the enzymes as a result of severe malnutrition (Carroccio A et al, Gut 1991; 32:796-799). [PubMed]
1942 Wolman IJ. Cystic fibrosis of the pancreas. Am J Med Sci 1942; 203: 900-905.
A really excellent review by Irving Wolman of Philadelphia (1905-1978),(Fig. 7) the editor of Clinical Pediatrics. He describes the “great catch basket of intestinal disorders comprised in the term “celiac disease” noting that “a new and significant entity has crystallized out” i.e. cystic fibrosis.
Wolman considers that Passini (1919 above) was the first to describe an instance of faulty digestion due to pancreatic disease. Parmalee (1935 above) related steatorrhoea to pancreatic deficiency as had Harper (1932 and 1938 above), Andersen (1938 above) and Rauch Litvak and Steiner in 1939 (above), all working independently, had succeeded to “throw this hitherto indefinite entity into sharp focus”.

Fig. 7. Irving Wolman. From US National Library of Medicine
– It should be noted that prior to Passini (1919), Byron Bramwell of Edinburgh, in describing an 18 year old boy whose diarrhoea responded to pancreatic extract he wrote, “came to the conclusion that the diarrhoea was due to defective metabolism in the upper part of the gastrointestinal tract: I suggest that it was probably due to disease or defective action of the pancreas”.
In his 1904 report Bramwell writes “I claim that there is a distinct variety or form of infantilism which is due to disease of the pancreas and that this, the pancreatic form of infantilism as I have ventured to term it, can be cured by administration of pancreatic extract. I consider this to be a distinct clinical entity – a disease which has not hitherto been recognised or described”.
1942 Attwood CJ, Sargent WH. Cystic fibrosis of the pancreas with observations on the roentgen appearance of the associated pulmonary lesions. Radiology 1942; 39:417-425.
The first paper on the chest X-ray findings in four patients with CF who had advanced pulmonary changes. Neuhauser et al, 1946 (below) later described the characteristic radiological changes in CF in two stages as follows – the first of emphysema or atelectasis and the second stage of infection. The former corresponds to the phase of partial or complete obstruction of the bronchi and bronchioles – clinically corresponding to the “early bronchitic phase” with which is associated a pertussis-like cough and precedes the onset of infection, destruction of the bronchial epithelium and bronchiectasis. In the “stage of infection” the hilar shadows are greatly accentuated even to a degree which may obscure the cardiac outline and give the appearance of a fluffy circumcardiac halo. Neuhauser also described shadows in the lung fields characteristic of peribronchial infiltration – often confluent suggesting pneumonia.
– The characteristic repetitive exhausting “pertussis-like” cough mentioned by these authors will be familiar to all parents and clinicians who have seen infants with CF who have significant chest involvement.

Normal chest X-ray

Typical advanced changes of cystic fibrosis
1942 Snelling CE, Erb IH. Cystic fibrosis of the pancreas. Arch Dis Child 1942; 17:220 – 226. (“Under the direction of Alan Brown MD FRCP Dept. of Pathology, Hospital for Sick Children, Toronto”).
– One of a number of reports around this time of children with CF diagnosed retrospectively by reviewing previous autopsy material in the light of the recent description of fibrocystic disease of the pancreas by Andersen in 1938.
In this reprot 19 cases were recognised from a review of autopsy material obtained in Toronto over the previous 20 years. All had physical signs of lung involvement in life such as bronchitis, collapse and consolidation of the lung, bronchopneumonia, and bronchiectasis on admission or during the period of observation. Classical pulmonary and pancreatic findings were described and there was considerable discussion on the respiratory epithelial metaplasia – “loss of ciliary activity results in a sort of physiological obstruction to the removal of secretion”. Epithelial metaplasia had been stressed by Andersen in her original 1938 paper and for some time was attributed to vitamin A deficiency.

Obituary in the Toronto Daily Star Sept 1960
Digression – The suffix “Under the direction of Alan Brown MD FRCP Dept. of Pathology, Hospital for Sick Children, Toronto” seems rather unusual and deserves comment.
Alan Brown (1887-1960) was a very distinguished and obviously an influential physician and pathologist. In 1915 as a young immigrant physician he joined the neonatal unit of the Hospital for Sick Children in Toronto. Apparently he was appalled by the high mortality and focused his attention on infant feeding practices. He held that feeding babies cows’ milk was dangerous, noting that “cow’s milk is for calves.” He urged mothers to breast feed their babies and insisted on pasteurizing the milk used in the hospital. But pasteurization was not law in Ontario, so many children were still being admitted to the hospital with bovine tuberculosis. But the politics of rural Ontario stood in the way of public health progress: a strong farm lobby staunchly opposed pasteurization on the basis of its cost and no elected politicians dared act.
By the mid 1930s, the tuberculosis problem was even more acute. In 1937 Dr. Brown took a stand and invited the Premier, Mitchell Hepburn, to visit the hospital to witness the rows of children suffering from the ravages of the disease caused, as he noted, by drinking raw milk. These children would not be there if the government would only act. The premier was duly impressed but the political opposition was strong and even the premier’s caucus was in revolt.
However, early in 1938, Hepburn met with a delegation of farmers opposed to pasteurization. They questioned evidence that untreated milk caused the disease but here, political skill, and a measure of luck, intervened. The premier recognized one of the farmers from earlier campaigning, and asked after his wife and children. “How many children do you have?” “I have five,” replied the surprised farmer. “But didn’t you have seven?” “Yes, but two died.” Sensing the truth, Hepburn went for the jugular: “They died of bovine tuberculosis, didn’t they? They drank milk from your own cows and died?” Hepburn persisted. “You came here today to protest against the pasteurization of milk. You have already lost two children to bovine tuberculosis, but that doesn’t prevent you from coming here to ask this Government to withdraw its bill and leave your children and other children open to the threat of death. What kind of man are you?”
Ultimately, Premier Hepburn’s tenacity won the day, illustrating the eventual impact of advocacy from a determined physician such as Dr Alan Brown who did not only work on the tuberculosis and pasteurization issue. During the 1920s, he established a research team to develop improved feeding methods for sick infants. This led to the invention of the first mineral-enriched baby cereal, Pablum, in 1930, developed jointly with Drs. Fred Tisdall and Theo Drake. For many years the royalties from sales of Pablum financed research at Sick Kids hospital (from www.med.uottawa.ca/sim/data/Public_Health…/Dr_Brown_e.htm)
 Dr Alan Brown was obviously a forceful character and subject of a later biography by Allison B Foster Kingsmill. “Dr Alan Brown:Portrait of a Tyrant(Canadian Medical Lives)”.
Dr Alan Brown was obviously a forceful character and subject of a later biography by Allison B Foster Kingsmill. “Dr Alan Brown:Portrait of a Tyrant(Canadian Medical Lives)”.
The author writes – Returning veterans, impatient to become doctors after their years of service in the armed forces, were not the immature students Dr. Alan Brown was accustomed to teach. Reactions to his threats to lock out any who were late for his class produced a famous anecdote which was a measure of his methods and a surprise to his veteran students. He accomplished his goals in unusual ways – sometimes cruel. The tales told by his staff and his students are revealing, often amusing and occasionally shocking. Over thirty years in the public eye he was revered by many and respected by all, including those in government, education and the business community.
1943 Farber S, Shwachman H, Maddock CL. Pancreatic function and disease in early life. I. Pancreatic enzyme activity and the celiac syndrome. J Clin Invest 1943; 20:827-833.[PubMed]
One of a series of important papers from Boston by Sidney Farber (figure 3) around this time discussing the differentiation between coeliac disease and cystic fibrosis – largely based on the enzyme content of the duodenal fluid to document and extend the conclusions of their preliminary report (Farber & Maddock, Am J Pathol 1941; 17:445). The authors used data from over 150 children – either normal controls or children with various nutritional disturbances, idiopathic coeliac disease or pancreatic fibrosis.
– The differentiation of pancreatic fibrosis from coeliac disease had taken on a new importance since the description of CF in 1938 as the prognosis for the two conditions was very different. They suggested differentiation of children with gastrointestinal features of coeliac syndrome into those with normal pancreatic function and those with abnormal pancreatic function – the later being a permanent and eventually fatal condition. Pancreatic achylia was a constant feature of those subsequently found to have pancreatic fibrosis at autopsy.
There was a clear separation of those with and without pancreatic fibrosis. So measurement of the pancreatic enzyme activity was considered mandatory for diagnosis of pancreatic fibrosis. Removal of 70-80% of the pancreas reduced the enzyme activity but did not produce pancreatic insufficiency (a finding later confirmed by DiMagno et al, 1973 below). Also Andersen (Am J Dis Child 1942; 63:643 above) had suggested duodenal trypsin as a reliable test for differentiating CF from coeliac disease.

Harry Shwachman
This was one of Harry Shwachman’s earlier papers as co-author. He would become one of the leading authorities on CF for many years to come. Shwachman (Fig. 9) received his BS degree from Massachusetts Institute of Technology in 1932. He qualified in medicine in 1936 at Johns Hopkins and subsequently did internships and residency at the Infant’s and Children’s Hospital Boston where he was assistant to Sydney Farber, the pathologist. Shwachman recalls “I was given the job of organising and running the clinical laboratories when I came back from the army (in 1946) by Dr Sidney Farber who was my chief at that time and he remained my chief as long as I’ve been at the hospital until my retirement in 1976” (from Fanos JH, 2008 below).
After various appointments in pathology, paediatrics and nutrition Shwachman founded the first specialised unit for CF at the Children’s Medical Center in Boston. He was active for 40 years with numerous publications and more than 300 papers. In 1958 with Kulczycki (below) he published their well known clinical scoring system. Also in 1964 with Louis Diamond and others he described the “syndrome of pancreatic insufficiency and bone marrow dysfunction” which was distinct from CF and came to be known as Shwachman’s syndrome or Shwachman-Diamond syndrome. In 1965 Shwachman started the Cystic Fibrosis Research Foundation. He became Professor of Pediatrics at Harvard in 1966.

Douglas Holscaw – author’s photo
Dr Douglas Holsclaw (fig. 10) of Philadelphia, who worked with Shwachman for some time, gave a very interesting and detailed tribute to him at a European CF meeting. He recalls that Shwachman’s first paper was on the bacterial content of swimming pools in the Boston Area. He was a complex man and a family man. Also, in addition to his work as a clinician, he was director of the clinical laboratory for many years. In 1950 he received the Mead Johnson prize for outstanding research and in 1976 a lifetime award for clinical work. He was always keen to become involved in the international area and was always on the phone to colleagues and to patients’ families – he maintained most situations could be managed with telephone advice. Shwachman made pioneering contributions in the area of cystic fibrosis. He was initially influenced by Sydney Farber in Boston to whom is usually accorded the first use of the term “mucoviscidosis”; but Douglas Holsclaw considers Shwachman was very influential in developing Farber’s concept of CF as a generalised disorder affecting the secretions in many organs.
Shwachman encouraged Gross, the paediatric surgeon to treat meconium ileus when the outlook appeared hopeless; he recognised there were various degrees of pancreatic insufficiency; described nasal polyps; developed the plate test for raised sweat chlorides – noted problems with the test after eating salted peanuts; incomplete expression of CF; developed the Shwachman-Kulczycki scoring system; reported the benefits of lung resection and recognised the association of rectal prolapse, male infertility and massive haemoptysis.
Shwachman would say of symptoms – “We do not have to put up with this!” He would frequently show “before and after” slides to everyone maintaining “we can make a difference”. Other favourite sayings were “Never assume anything”. “A good manuscript always requires 9 drafts”. ”You can never have too much data – you never know when you’re going to need it”. He trained at least 55 people from over 26 countries – the list reads like a “Who’s Who” of CF. His name has been spelt incorrectly repeatedly – Douglas Holsclaw has seen 20 different versions!! The legendary CF physician Warren Warwick of Minnesota regarded Harry Shwachman as a great star of CF and observed – “Harry Shwachman had an absolute memory for everything he had seen and made many astute clinical observations that I wish I could remember today. If he had a weakness it was not being able to make the best judgments about experimental studies”.
There was an interesting interview with Harry Shwachman and his wife in 1984, two years before his death, which was published in 2008 and makes really fascinating reading (Fanos JH. “We kept our promises”: An oral history of Harry Shwachman, MD. Am J Med Genet A. 2008; 146A:284-293). [PubMed].
1944 May CD. Fibrosis of the pancreas in infants and children. Proc R Soc Med 1944; 37:311-313. [PubMed]

Charles May medicine.uiowa.edu
This was probably the first published presentation on CF in the UK – Charles May was in London and a major in the US Army Medical Corps at the time (Blackfan & May, 1938 above). This is a beautifully succinct paper describing the 35 patients (from no less than 2800 paediatric autopsies in Boston) who had the typical pancreatic lesions of CF, presumably including those described by Blackfan & May, 1938 (above), and 25 additional examples.
May writes “Almost all the exocrine portion of the gland is profoundly affected. The acini and small ducts are greatly dilated by an inspissated secretion. Frequently the lumina of the ducts and acini are so markedly dilated that the appearance in that of a cystic structure but there are no true cysts. Extensive fibrosis is found. The Islets of Langerhans are not involved: they appear and function quite normally. On the other hand the damage to the acini is so severe and widespread that normal production of enzymes by the pancreas is inconceivable”.
No child in the series had lived longer than 15 years. Staphylococcus aureus was usually the sole organism with no response to sulphonamides – which had been available from the late Thirties. May observed prophetically “Perhaps penicillin will afford a more successful approach” – but this was not available until the mid-Forties when it certainly did have a dramatic effect according to di Sant’Agnese (di Sant’Agnese 1946 below); however, Shwachman later recalled he was less impressed (Fanos JH. 2008. below). Charles May observed that “Ultimate recovery of these patients will depend on recovery from and prevention of the pulmonary infection”. This latter was a useful observation and certainly was not stating the obvious, as it may seem today, for CF was still considered by some experts, including Dorothy Andersen herself, as primarily a gastrointestinal pancreatic disorder with secondary nutritional consequences, particularly vitamin A deficiency, causing a variety of systemic problems one of which was the chronic respiratory infections.

Fig. 11. Donald Paterson
1943 Donald Paterson diagnosed the first child with CF at the Hospital for Sick Children, Great Ormond Street, London (according Dr Martin Bodian and Dr Archie Norman).
Dr Donald Paterson (1890-1969) (Fig. 11) was a Canadian who trained in medicine at Edinburgh. In 1919, after First World War Service, he became a house surgeon and later consultant at The Hospital for Sick Children, Great Ormond Street, London. For many years, until he returned to Canada in 1947, Paterson was a leading figure in British paediatrics and with James Spence and Frederick Still founded the British Paediatric Association (now the Royal College of Paediatrics and Child Health) in 1928 (also Patterson et al, 1945 below).
The 1945 paper reviews Patterson’s successful experience in 1943-44 in treating 26 children with coeliac disease with a regimen similar to that advocated by May et al, (J Pediatr 1941; 21:17) with liver extract and vitamin B complex. The author mentions that, in the investigations of children with coeliac disease, fibrocystic disease of the pancreas was excluded by aspiration of the duodenal contents as recommended by Andersen and Early (Am J Dis Child 1942:63:891 above).
1943 Shohl AT, May CD, Shwachman H. Studies of nitrogen and fat metabolism on infants and children with pancreatic fibrosis. J Pediatr 1943; 23:267-279.
A study of nine patients with cystic fibrosis. The striking feature of the infants with CF was the excess of nitrogen found in the faeces when normal diets were eaten, the patients remaining in negative nitrogen balance. Average weight of dried faeces/day – Normal infants 7.3 g – CF infants 33.4 g. Average nitrogen content of faeces/day – Normal infants 0.39 g – CF infants 1.7 g. Average fat content of dried faeces/day – Normal infants 3.1 g – CF infants 16.8 g. On a fat-free diet of casein hydrolysate and glucose, nitrogen retention was in the normal range and excretion normalised.
The authors concluded that hydrolysed casein could be utilised in contrast to whole protein. Subsequently feeds containing hydrolysed protein (e.g. Pregestimil) were shown by others to be useful in improving malabsorption in some infants with CF where control of the malabsorption had proved difficult – particularly after meconium ileus operations and with cow’s milk intolerance.
– The concept of an “elemental diet”, which required less digestion, to improve absorption was also later pioneered by Dr Jimmy Allen, a general paediatrician in Macclesfield, England (Allan JD et al, 1970 and 1973 below) and created considerable interest in the Seventies. Prior to the introduction of the acid resistant microsphere pancreatic enzyme preparations Pancrease and Creon in the early Eighties, it was almost impossible to maintain a normal nutritional state as the children became older and their chest infection became progressively worse.
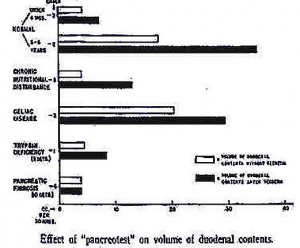
Fig. 12. Effect of “Pancreotest” on the volume of duodenal contents.
1943 Maddock CL, Farber S, Shwachman H. Pancreatic function and disease in early life. II. Effect of secretin on pancreatic function of infants and children. Am J Dis Child 1943; 66:370-375.
These workers from Boston noted that the hormone secretin (discovered by Bayliss and Starling in 1902) had been used to stimulate pancreatic secretion only in adults up to that time. So they performed 26 secretin stimulation tests on 21 young patients and found trypsin was absent from the duodenum after intravenous stimulation by secretin in the 7 children with cystic fibrosis. They noted that in non-CF children “the increased flow of duodenal juice that followed the injection of secretin occurred in 3 – 5 minutes. Often the pancreatic flow was so copious that the bile coloring of the duodenal fluid was diluted so that fluid was colourless by the end of twenty minutes at which time the flow rate diminished. The most striking feature was the small volume of pancreatic juice obtained in patients with pancreatic fibrosis and the apparent failure of secretin to stimulate pancreatic flow in these patients in the “pancreatic fibrosis” group”. (figure – Pancreatic fibrosis is the lower most pair of bars; the open and closed bars are respectively the volumes of duodenal contents before and after secretin. The x axis is “ccs per 30 mins”).
– More extensive studies on pancreatic function in CF with secretin and pancreozymin were later reported by others – notably by Hadorn et al, 1968 (below) and these are usually quoted when discussing the subject; even more recently the subject was studied by Kopleman et al in Toronto (N Eng J Med 1985; 312:329-334 below). All have found markedly reduced fluid and bicarbonate output in all patients with CF and, in the majority, a markedly reduced enzyme output.
1943 Shwachman H, Farber S, Maddock CL. Pancreatic function and disease in early life. III. Methods of analysing pancreatic enzyme activity. Am J Dis Child 1943; 66:418.
This is a description of the two methods of measuring tryptic activity – the tube method and gelatin method. One of series of papers with Sydney Farber regarding the investigation of pancreatic function.
1943 Farber S. Pancreatic insufficiency and the celiac syndrome. N Engl J Med 1943; 229:653-657. [PubMed]
Sydney Farber from the Infants Hospital and Children’s’ Hospital, Boston, described the gradual recognition in the literature of a group of infants with coeliac disease who also had pancreatic lesions. He suggested, as had Wolman in 1942 and others, that previously Parmalee (1935) had made this clear distinction when reviewing coeliac children with pancreatic lesions.
Farber postulated CF was a generalised disease – obstruction by thick mucus – “More important than infection is the obstruction (in the airways) caused by the tenacious mucus comparable in consistence with the viscid pancreatic juice. Inspissation of secretions and dilatation of glandular structures of the same general character as these (pancreatic) alterations in the acini and small ducts of the pancreas are found at autopsy on these patients in the glands of the trachea, bronchi, duodenum, gall bladder and intestinal tract”.
Farber is credited with introducing the term “mucoviscidosis” (Farber 1942 above) perhaps, according to Douglas Holsclaw, influenced by his clinical colleague Harry Shwachman.
1944 Howard PJ. Familial character of fibrocystic disease of the pancreas. Am J Dis Child 1944; 68:330-332.
Philip Howard of Detroit reviewed the previous twelve instances of fibrocystic disease of the pancreas where a familial occurrence had been reported including instances where both or only one of twins had been affected. He reported an unusual number of infant deaths in two families in which proven CF existed and suggested the family furnished an instance of heterozygous inheritance (see Andersen and Hodges, 1946 below).
– This is the first suggestion that CF was an inherited disorder – later this was established on a larger series of children with CF by Andersen and Hodges (1946 below).
1944 Menten ML, Middleton TO. Cystic fibrosis of the pancreas: report of 18 proved cases. Am J Dis Child 1944; 67:355-359.
Another paper based on a search through previous paediatric autopsies revealed 18 cases of CF from The Children’s Hospital, Pittsburgh; all had diffuse definite dilatation of the pancreatic acini and associated pneumonia. The authors state that radiological examination of the lungs showed a remarkable similarity in the unusual enlargement of the hilar shadows and a bilateral shadow decreasing to the periphery and suggest this as a diagnostic feature. Three had bronchoscopy during life – in one showing a normal trachea and in two reddened thickened mucous membrane containing thick mucopurulent material.
– This is the first mention of the bronchoscopic appearances seen in cystic fibrosis.
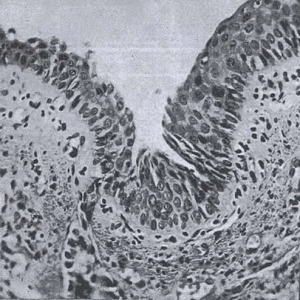
ig. 13. Squamous metaplasia of respiratory epithelium. From Philipsborn et al 1944 with permission.
1944 Philipsborn HF Jr, Lawrence G, Lewis KC. The diagnosis of fibrocystic disease of the pancreas based upon 26 proved cases. J Pediatr 1944; 25:284-298.
Yet another retrospective review from Chicago of previous paediatric autopsies to identify those with characteristic pancreatic findings of cystic fibrosis. The clinical histories and pathology findings in 26 proven cases of CF since 1938 were reviewed noting that the diagnosis had seldom been made in life. The characteristic pancreatic findings of CF were present in 3.5% of all the children who had autopsies at the Children’s Memorial Hospital, Chicago. The authors suggested that the antemortem diagnosis of this not-so-uncommon condition can be facilitated by realising the various forms in which the disease manifests itself and by the judicious use of laboratory procedures including vitamin A absorption curves, duodenal drainage after intravenous secretin, mentioning that Ivy (Am J Dig Dis & Nutr 1936; 3:677) had noted reduced duodenal enzyme concentrations after secretin stimulation was significant. There is considerable detail including illustrations of the squamous metaplasia of the respiratory epithelium (figure) considered, by Andersen and others, to be related to vitamin A deficiency and a factor in perpetuating the respiratory infections.
Dr Herbert Philipsborn Jr (1915-2010) was a distinguished paediatrician who was on the staff of the Children’s Memorial Hospital for 55 years.
1944 Farber S. Pancreatic function and disease in early life. V. Pathologic changes associated with pancreatic insufficiency in early life. Arch Pathol 1944; 37:238-250.
Detailed description of the morphologic changes in multiple organs in 87 patients with CF. Farber noted all the changes in multiple organs were secondary to obstruction of mucus secreting glands. He suggested that CF was a generalised disease of exocrine glands with inspissation of mucus secreting glands and suggested the name “mucoviscidosis”.
1944 Fanconi G, Botsztejn A. Familial pancreatic fibrosis with bronchiectasis. Schweiz Med Wchnschr 1944; 74:85. (German).
Fanconi’s 1936 report (Fanconi et al, 1936 above) is considered by some Europeans to be the first clear description of cystic fibrosis. Here the authors described the family trees of 25 children with fibrocystic disease of the pancreas. They considered, quite incorrectly at it turned out, that recessive transmission was unlikely as there was consanguinity in only 3 of their cases and the proportion of affected siblings appeared too high. So they postulated the cause to be an early intrauterine toxic factor akin to haemolytic disease of the newborn.
– Andersen & Hodges, 1946 (below) were the first to correctly identify the Mendelian recessive mode of transmission, although a heterozygous mode had been suggested as likely by Howard in 1944 (above).
1945 Baggenstoss AH, Kennedy RLJ. Fibrocystic disease of the pancreas: Study of 14 cases. Am J Clin Path 1945; 15:64-70.
Another report of 14 infants with cystic fibrosis identified by review of previous autopsies performed over the past 18 years at the Mayo Clinic. Patients were classified into meconium ileus (1), respiratory (8) and coeliac (5). The pancreatic histology was typical of CF and the mean age at death was 8.5 months. In discussing the suggested possible causes (vitamin A deficiency, viral infection, and abnormal pancreatic secretion and obstruction of the pancreatic duct) they conclude that “the possibility that abnormally thick secretion causes intrinsic obstruction in the acini and small ducts must be seriously considered”.
1945 Patterson D, Pierce M, Peck E. The treatment of coeliac disease with vitamin B complex and concentrated liver. Arch Dis Child 1945; 19:99–110.
This paper, although not concerning CF directly, is included as Henderson 1945 (below) mentions an unsuccessful trial of this treatment in a child with cystic fibrosis. This paper reviews Patterson’s successful experience in 1943-44 in treating 26 children with coeliac disease with a regimen similar to that advocated by May et al (J Pediatr 1941; 21:17) with liver extract and vitamin B complex.
The author mentions that, in the investigations of these children, fibrocystic disease of the pancreas was excluded by aspiration of the duodenal contents as recommended by Andersen & Early (Am J Dis Child 1942:63:891) – a procedure by this time established as essential in the diagnosis of cystic fibrosis and differentiating CF from coeliac disease.
1945 Henderson WD. Cystic fibrosis of the pancreas. Arch Dis Child 1945; 20:179-181.
Henderson described a boy aged 22 months with CF describing the difficulties in differentiating from coeliac disease particularly on the amount of split fat (88.6% in this case) which could be misleading. The child developed measles and then bronchopneumonia that proved fatal. The diagnosis of CF was confirmed at autopsy and the histology of the pancreas was diagnostic. Treatment seemed hopeless but Henderson suggested that liver extract and vitamin B, as used in coeliac disease, might be worthwhile (Paterson et al, 1945 above) as a drop in the fat content of the stool was noted after seven injections. Sulphonamides and even penicillin, which was just becoming available, had no effect.
– This was one of the few CF publications from the UK at this time. Dr Douglas Henderson was “Honorary Physician” to Woking Victoria Hospital. This was just before the start of the National Health Service in 1948 when many medical consultants in the UK were so-called “Honoraries” i.e. they gave their services free, or for a nominal retainer, to the hospital and relied on their private practice for income. As there was less private work in paediatrics than in the other main specialities there were very few full time paediatricians in the UK at that time.
1945 Wissler H, Zollinger HU. Familial congenital cystic fibrosis of pancreas with bronchiectasis. Helv Paediatr Acta 1945; 1 (Suppl 1):3-88.
This paper from Zurich describes the clinical and pathological features of CF noting that lung changes may be absent in “nurslings with pancreatic fibrosis who die early”. The cystic pancreatic changes often are only detected by microscopy and may be overlooked. “It seems highly probable that the inflammatory changes in the pancreas, biliary passages and bronchial walls are the result of a single process… probably due to intrauterine damage of maternal origin… no reasons for accepting a primary avitaminosis as the cause and a recessive hereditary disease is improbable”.
– These Swiss authors mistakenly considered an hereditary cause of CF to be unlikely and suggested the cause was more likely due to intrauterine damage as had a previous paper from Switzerland (Fanconi & Botsztejn, 1944. above).
1945 Andersen DH. Celiac syndrome. II. Fecal excretion in congenital pancreatic deficiency at various ages and with various diets, with discussion of optimal diet. Am J Dis Child 1945; 69:221-230.
Faecal fat was normal in 3 patients aged less than six months but increased in all patients above that age. Some infants are pancreatic sufficient for some months as was later confirmed by Kevin Gaskin et al in Australian screened infants with cystic fibrosis (Waters et al, 1990 below; Gaskin et al, 1991 below). Reduction of dietary fat resulted in proportionate decrease in faecal fat. Fat excretion was usually reduced by pancreatin but the effect was variable. Various dietary combinations were tried leading to the suggestion that the optimal diet was approximately as follows: “The protein should provide 25% of the calories: the proportion of fat should be small but eggs and fat soluble vitamins should be included: the carbohydrate should be provided in part as sugar and for older infants and children part of it may be in the form of cereal starches and potato if these foods are clinically tolerated”.
1945 Andersen DH. Celiac Syndrome. III. Dietary therapy for congenital pancreatic deficiency. Am J Dis Child 1945; 70:100-113.
Thirty of 38 children were diagnosed in life. Andersen recommended a diet low in fat, high in protein with liberal allowance of fruit and vegetables and moderate restriction of starch. Precise details of the dietary constituents are listed – obviously a very low fat diet. The importance of supplementary vitamin A was stressed. Prognosis was better if dietary treatment started before the onset of “suppurative bronchitis” – if started later it would prolong life but would not alter the bad prognosis. In fact the prognosis was terrible at that time at whatever time the dietary treatment was started but the view expressed here is still reflecting Andersen’s lingering belief that prevention of the respiratory epithelial changes due to vitamin A deficiency would ward off the chest infections.
– Dorothy Andersen was one of the first to recognise the damaging effects of viral “colds” and recommended isolation of infants, even from the family, during the hazardous first year – after this it seemed wiser to risk respiratory tract infection in the interest of providing the child with normal human contacts. The extremely deleterious effects of viral infections particularly in young infants with CF have been confirmed by numerous authors (Smyth AR et al. Effect of respiratory virus infections including rhinovirus on clinical status in cystic fibrosis. Arch Dis Child 1995; 73:117-120.[PubMed]).
These valuable clinical observations from Dorothy Andersen are interesting as she was a primarily a pathologist; apparently the New York children were under the care of Paul di Sant’Agnese as Milton Graub recalls “since Dr Andersen was a pathologist she referred Lee (his son) for further clinical care to Dr Paul di Sant’Agnese”.
Dr Margaret Harper, the Sydney paediatrician, clearly saw Dorothy Andersen as a pathologist! She commented “Andersen’s article from the pathological laboratory of the Babies Hospital New York reported 49 cases (of CF) culled from various sources; it is written from the pathological standpoint without personal clinical experience of the disease”! This was, of course, true as all the information in the article at the time it was written was gleaned from autopsy reports.
1946 West CD, Wilson JL, Eyles R. Blood amino nitrogen levels: changes in blood amino nitrogen levels following ingestion of protein hydrolysate in infants with normal and with deficient pancreatic function. Am J Dis Child 1946; 72:251-273.
The absorption of whole protein and casein hydrolysate was compared. There was a significantly smaller rise in blood amino acids after a whole protein meal in infants with CF but similar increases in blood amino acids after casein hydrolysate in both normal and CF children. Authors suggested that substitution of the hydrolysate for whole protein should benefit CF children.
– This was further confirmation of the suggestion of Shohl et al, 1943 (above) and subsequently shown to be very useful in practice (Allan et al, 1970 & 1973 below and the “Allan diet”) that protein hydrolysate was better absorbed than whole protein by children with cystic fibrosis.
1946. Farber S. Some chronic diarrhoeal disorders in early life. Chic M Soc Bull 1946:49:79-83
Sydney Farber discusses the aetiology of chronic diarrhoea of infants including bacterial infection, parasites and even ulcerative colitis but mainly discusses the celiac syndrome, the four common causes being coeliac disease, chronic parenteral infection, congenital malformations and pancreatic insufficiency – the last caused by congenital malformations, healed pancreatitis or in older patients a pancreatic stone or neoplasm. He mentions that rarely vitamin A deficiency producing “metaplasia and epithelium piling up of keratinised debris in the epithelial lined spaces with obstruction of the pancreatic duct”. However, Farber did again, as he had on previous occasions, emphasise the generalised nature of the condition rather than it being a pancreatic disorder with secondary nutritional consequences. “Most infants and children with the clinical picture of pancreatic insufficiency show involvement of many other structures including salivary glands, upper respiratory tract, lungs, gall bladder, bile capillaries intestinal tract and pancreas. The change appears to be an alteration of the physical character of the gland secretion” – this became accepted as Farber’s concept of the disease as he repeatedly emphasised this point.
1946 Di Sant’Agnese PA, Andersen DH. Celiac Syndrome IV. Chemotherapy in infections of the respiratory tract associated with cystic fibrosis of the pancreas; observations with penicillin and drugs of the sulphonamide group, with special reference to penicillin aerosol. Am J Dis Child 1946; 72:17-61

ig. 15. Paul di Sant’Agnese. With permission of Dr Carl Doershuk
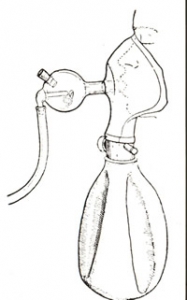
Fig.14. Nebuliser used for administration of penicillin. From Am J Dis Child,1946.
The first report of the use of penicillin in cystic fibrosis. In 1943 a small quantity of penicillin had become available from the US Army to treat three children with CF and a further 2 in 1944 with intramuscular penicillin; the results were variable. Bryson and co-workers at the Carnegie Foundation were first to investigate the use nebulised penicillin (Bryson V et al, Science 1944; 100:33). In 1944 Alvan Barach of the Presbyterian Hospital was using a penicillin aerosol to treat asthma and bronchiectasis (Barach et al, Ann Int Med 1945; 22:485). Similar apparatus was used by di Sant’ ‘Agnese for these children with CF; it consisted of a rubber mask with re-breathing bag and nebuliser with a flow of oxygen into the nebuliser (Fig. 14). This is an interesting first report describing early treatment in CF and in particular the first use of nebulised penicillin 20,000 units seven times daily with or without intramuscular penicillin 160,000 units daily. According to di Sant’Agnese, dramatic response occurred in 15 infants and young children with CF who eventually received the treatment but little success was achieved in infants less than a year old.
– From 1947 there were numerous publications on the use of the recently available penicillin in other conditions by various routes both oral, by aerosol and even powdered inhalations – no less than 18 such publications were reviewed in The 1948 Year Book of Pediatrics. (Poncher HG (ed). Year Book Publishers. Chicago).
Undoubtedly a new era of treatment of infection had begun.
Paul di Sant’Agnese (1914 – 2005) (Fig 15) wrote that “Penicillin is the first drug known to affect the course of fibrocystic disease after cyanosis has marked the existence of suppurative Staphylococcal bronchitis”. Later, in 2001, he recalled “In most patients the results (of penicillin treatment) were dramatic. From death’s door, slowly dying from chronic pulmonary disease while we watched helplessly, patients revived in a few days”. Sulphonamides were said to be useful in prophylaxis and for intercurrent infections but not after the stage of suppurative bronchitis.
Dr Lynn Taussig, who worked with di Sant’Agnese, later recalls that Paul di Sant’Agnese and Harry Shwachman were definite rivals but had considerable respect for each other. Di Sant’Agnese came from a noble family in Italy. His father was an obstetrician and radiotherapy expert who was physician to the Italian Royal Family. Paul went to Rome medical school and then came to USA in 1939 to study medicine in New York. He was chief resident in paediatrics and published his first paper on tick paralysis – as initially he worked in infectious disease and immunology; he studied the effect of immunisations in early life.
He went to work with Dorothy Andersen at the Presbyterian Hospital in New York. In 1950 he described glycogen storage disease of the heart (Pompe’s disease) and as a gastroenterologist in the early 1950s he wrote on coeliac disease.
In 1953 di Sant’Agnese’s observation of abnormal sweat electrolytes in CF was one of the most important observations in the history of CF and provided the basis of the sweat test (Di Sant’Agnese et al, 1953 below). He wrote over 140 papers mostly on CF – physicochemical differences, mucoproteins, calcium in secretions, and with West the first report of pulmonary function tests, pneumothorax, ventilation, pancreas, duodenal contents, and measurements of absorption. He was the first to describe the liver changes – a few months before Shwachman’s group. In fact, he had an interest in every aspect of the condition.
But the patient always came first and he was an outstanding clinician as well as a gifted researcher. In 1955 he was involved with the medical aspects of the CF Foundation. In 1960 in Europe he met Archie Norman, David Lawson; the International Cystic Fibrosis and Mucoviscidosis Association (ICFMA) had 28 delegates and guests from 14 countries.
Di Sant’Agnese said he was most proud of training so many associates and colleagues in CF care and research. “If your plan is for one year, plant rice, if for 10 years plant trees, if for life – educate!”
– In 1946 this present paper on the use of penicillin for fibrocystic disease, with special reference to aerolised penicillin, was the first publication on the use of this new antibiotic in cystic fibrosis.
1946 Wigglesworth FW. Fibrocystic disease of the pancreas. Am J Med Sci 1946; 246:351.

Fig. 16. F W Wigglesworth
This paper lent authoritative support to Farber’s theory that CF was a systemic disorder characterised by the production of an abnormally viscous secretion with secondary effects in many organs. The author (Fig.16) surveyed the literature and noted that the secretions found in the pancreas and the mucous glands did not appear to be the same. Wigglesworth concluded that it was not necessary to attribute too much importance to the pancreatic lesions as a cause of the general changes (as was still suggested as a possibility by Dorothy Andersen) and that Farber’s theory of widespread involvement of mucus secreting glands was perhaps the most important of theories of aetiology but still required to be proved.
1946 Bodian M. Fibrocystic disease of the pancreas. Arch Dis Child 1946; 21:179.

Fig. 16a Martin Bodian
Martin Bodian (fig. 16a) was pathologist at the Hospital for Sick Children, Great Ormond Street, London. In this first short report on CF to a meeting of the British Paediatric Association, Bodian reviewed 30 children with CF seen at the Hospital for Sick Children, Great Ormond Street, London between 1943, (when the first case there was diagnosed by Donald Patterson), and 1946.
Bodian also suggested that there was evidence for CF being a pluri-glandular disease with widespread abnormal secretions which inspissates in the pancreatic ducts and acini followed by atrophy and fibrosis. The measurement of trypsin in the duodenal contents was the most useful investigation. Clinically the patients were grouped as neonatal intestinal obstruction (3 cases), pneumonia in infancy (13) and coeliac syndrome (14).
The details of the patients are included in his 1952 book (Bodian, 1952 below for details).
1946 Kennedy RL. Cystic fibrosis of the pancreas. Nebr Med J 1946; 31:493-496.
This is a detailed report of experience from the Mayo Clinic. The outlook for children with CF was still terrible and 18 of 28 (64%) of patients in the series died aged less than seven years. Treatment was with vitamin A 100,000 IU one or more times per week, low fat high protein diet, pancreatin granules and penicillin inhalations. There is an interesting discussion of current theories of pathogenesis – the suggested possibilities being 1). Desquamation of pancreatic ducts due to vitamin A deficiency. 2). Abnormal pancreatic secretion similar to inspissated secretions noted in other organs indicating a generalised disturbance and 3). Infection as the main cause.
– There was a previous report by Kennedy & Baggenstoss (Proc Mayo Clin 1943; 18:487) outlining the main features of steatorrhoea, chronic cough and absence of trypsin in the duodenum.
1946 Palmer HD, Danielson WH, Lubchenco LO, Binkley EL. Absorption of vitamin A following enteral use of prostigmine in cystic fibrosis of pancreas. Am J Clin Path 1946; 16:535-549.
Prostigmine was apparently used by Flack to improve bowel motility, which was presumably the rationale for this study. However, oral or parenteral prostigmine did not improve vitamin A absorption in 3 children with CF but did induce active intestinal peristalsis. So the authors concluded that prostigmine did induce active peristalsis in patients with cystic fibrosis and seemed to promote regularity of bowel movements but the improvement was not greater than in children receiving their current supportive therapy.
1946 Andersen DH, Hodges RC. Celiac syndrome V. Genetics of cystic fibrosis of the pancreas with consideration of the etiology. Am J Dis Child 1946; 72:62-80.
Investigating 47 of their own families and 56 from the literature, the authors concluded that the familial incidence indicated a hereditary disorder with a recessive mode of inheritance, but which required more than one factor for its expression. Although the incidence in siblings approximates to the 25% expected of a Mendelian recessive trait, an hereditary condition requires more than one factor for its expression.
– This was a quite different explanation to that given by Fanconi et al, 1944 but in line with that of Bodian, 1952 (below). Andersen continued to believe that “the pulmonary infection is the result of the nutritional deficiency” – obviously she considered this to be the additional factor required.
This is the first clear statement, backed by clinical evidence, that CF is inherited in a Mendelian recessive manner. Others, including David Lawson, would also speculate on the relative contribution of the basic defect and the secondary effects resulting from the defect (such as chest infections and malnutrition) to the ultimate outlook for the patient. i.e. if the secondary effects could be prevented would the outlook be much better? This was not clear at this time as the outlook was so poor. Subsequent progress showed that this was indeed the case in view of the steady improvement in outlook that occurred with treatment of the secondary effects before any modification of the basic defect was achieved.
1946 Neuhauser EBD. Roentgen changes associated with pancreatic insufficiency in early life. Radiology 1946; 46:319-328.

Fig. 16b Edward Neuhauser & John Caffey 1975. Society of Pediatric Radiologists
Radiological changes described were an increased lung volume causing flattening of the diaphragm, increase in radio-translucency and widening of the intercostal spaces. There was frequent lobular atelectasis and obstructive emphysema. Later there was accentuation of the hilar shadows and bronchovascular markings with irregular emphysema, increased hilar markings atelectasis and pneumonia in the terminal stages (also Atwood & Sergent, 1942 above).
1947 Fanconi G, Botsztejn A. Special forms of cystic fibrosis with bronchiectasis. Helv Paediatr Acta 1947; 2:279-288.[PubMed]
A number of children where the diagnosis had been disguised by special features including oedema in six children. One child who died at six weeks had a large liver, hyperglycaemia and glycosuria. At autopsy the pancreas showed evidence of fibrocystic disease and the liver of diffuse cirrhosis.
1948 Pitt D. Fibrocystic disease of the pancreas; a review of 14 cases. Med J Australia 1948; 1:91-100.[PubMed]
Pitt described 14 patients with CF from the Children’s Hospital, Melbourne, Australia, two of whom, aged nine and 10 years, had multilobular portal cirrhosis of the liver. He noted several case reports had appeared “since Passini described his case in 1919” (above). In 1946 Blaubaum reported 2 cases from the Children’s Hospital Melbourne (Blaubaum PE. Med J Australia 1946; 1:833-837) and now these are a further 12 cases, 9 of whom had died. The paper is a detailed report of the condition which the author regarded as “essentially a deficiency disease, the deficiency being the external secretion of the pancreas, would seem to provide grounds for optimism. Replacement of the defective element may well be as effective in this as in other deficiency diseases”.
– This is an extremely detailed paper and a worthy predecessor of the subsequent CF work from Melbourne and which ultimately, when contrasted with the inferior UK results in the Seventies (Phelan & Hey, 1984 below), was to have a major influence on the care of people with CF in the UK. However, in the present paper there is still the rather too simple implication that if the pancreatic secretion were to be replaced, the other manifestations of the condition would be controlled, which was not the case.
1948 Atkins JP. Bronchoscopic observations on the pulmonary aspects of fibrocystic disease of the pancreas. Ann Otol (St Louis) 1948; 57:791-801.[PubMed]
An early report of bronchoscopy in children with CF using the rigid bronchoscope – the only type available at that time. Atkins stated the expiratory intrusion of the posterior wall into the lumen produced crescenteric cross section of the bronchi not differing from that seen in bronchial asthma.
– Prior to this, Menton & Middleton (1944 above), reporting 18 children with CF from Pittsburgh in 1944, incidentally described the bronchoscopic appearances in three patients who were so examined. Later Robert Wood in the USA pioneered the use of flexible bronchoscopy in children from 1975 onwards (See Wood et al, 1973 & 1975 below).
1948 Hiatt R, Wilson P. Celiac Syndrome VII. Therapy of meconium ileus, report of 8 cases with review of the literature. Surg Gynec Obstetr 1948; 87:317-327.[PubMed]
These were early days for paediatric surgery and there were very few specialist paediatric surgeons and anaesthetists. In this first report of survival of infants with meconium ileus the surgeons at the Babies Hospital, New York irrigated the bowel via an incision at the junction of the dilated bowel and the portion containing inspissated meconium, and then closed the bowel. The operation was successful in 5 of 8 cases – better results than expected and very good for the time.
– Later in 1955 Shwachman comments that prior to this report the “the surgical correction of the obstruction was considered hopeless” and this was “the first important contribution to correction of this condition”.
1948 Sinclair W Jr. Cystic fibrosis of the pancreas. Ohio Med J 1948; 44:1024. [PubMed]
This paper is mentioned as the first report of partial pancreatic involvement in a later report by Gibbs et al, 1950 (below). Autopsy diagnosis of an infant aged five months with CF in whom approximately one half of the pancreas, and that mostly in the tail, appeared to be involved. The infant had a normal concentration of trypsin in the duodenal aspirate and a normal output of stool fat. The usual bronchitis and bronchopneumonia expected with CF were the cause of death.
1949 Lowe CU, May CD, Reed SC. Fibrosis of pancreas in infants and children: statistical study of clinical and hereditary features. Am J Dis Child 1949; 78:349-374. [PubMed]
These authors from the University of Minnesota reviewed the hereditary aspects of 134 patients with CF seen over 10 years at the Infant’s and Children’s Hospital, Boston where previously May had worked. There were 118 sibships and the authors concluded that the condition was determined by a recessive gene as had Andersen and Hodges (Andersen & Hodges, 1946 above). They noted good weight gain before first symptoms and fair gains thereafter in those who survived. In discussing the clinical features they noted 44 children showed osteoporosis and retarded bone age. They questioned the importance of vitamin A deficiency in the pathogenesis as had others.
– This is one of the earliest mentions of osteoporosis in people with CF – a complication that was to become a major problem for adults with CF in the years to come.
1949 May CD, Lowe CU. Fibrosis of the pancreas in infants and children; illustrated review of certain clinical features with special emphasis on pulmonary and cardiac aspects. J Pediatr 1949; 34:663-687. [PubMed]
A very comprehensive study of 134 patients from Minneapolis. Virtually all had early onset of signs of malabsorption, 29 had meconium ileus and all had pulmonary involvement Staphylococcus aureus being the usual organism. Cardiac failure was observed in six patients. The authors considered that aerosol antibiotic (penicillin) inhalation, recently described by di Sant’Agnese (1946 above) was the first substantial advance in treatment. The authors believed that “the pulmonary process begins soon after birth, is most intense between 6 to 18 months and then subsides somewhat”. Even improved nutrition did not seem to halt the progress.
1949 Shwachman H, Crocker AC, Foley GE, Patterson PR. Aureomycin therapy for the pulmonary involvement of pancreatic fibrosis (mucoviscidosis). N Eng J Med 1949; 241:185-192. [PubMed]
The first enthusiastic report of a favourable response of children with CF to the wide spectrum antibiotic aureomycin – indeed Shwachman was the first to use this broad spectrum antibiotic in cystic fibrosis. Aureomycin had been used from September 1948 –“the substitution of an orally effective antibiotic such as aureomycin for the expensive and more difficult form of aerosol therapy has obvious merits”. Nebulized penicillin and streptomycin were routine treatments at the time. Thirty five children were treated with aureomycin for up to 4.5 months with excellent results in 31 and most of the Staphylococci remained sensitive. However, diarrhoea was a frequent complication and “the appearance of Proteus vulgaris, Pseudomonas aeruginosa and fungi overgrowing or replacing the initial flora” were observed repeatedly “reminiscent as seen after prolonged penicillin and streptomycin therapy”.
– So already prolonged use of an antibiotic was resulting in a change of the bacterial flora from S. aureus to gram negative organisms including Pseudomonas aeruginosa. In the years to come there would be protracted discussions as to whether long term flucloxacillin (an anti-staphylococcal antibiotic) increased the incidence of Pseudomonas infection. It probably does but that is no reason for allowing S. aureus to flourish and damage the lungs as 1949 still seems to be occurring! It seems as if any organism is eliminated or specifically prevented from infecting the CF airways, another will step in. Hence the sequence S. aureus -> P. aeruginosa -> Aspergillus or S. maltophilia etc. So each has to be treated aggressively and eradicated as soon as it appears.
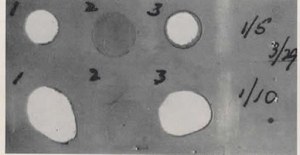
Fig. 17. A photograph in the paper (figure) shows gelatin film acted upon by stool suspensions from three individuals (1, 2 and 3). Concentrations of 1/5 on upper row and 1/10 are on the lower row being the more dilute.
1949 Shwachman H, Patterson PR, Laguna J. Studies in pancreatic fibrosis: Simple gelatin film test for stool trypsin. Pediatrics 1949; 4:222-230. [PubMed]
In this paper previous work on stool trypsin was reviewed, even as far back as the first duodenal catheterisation by Hess AF (Am J Dis Child 1912; 4:205). Faecal suspensions were placed upon unexposed and unfixed gelatin film and the extent of removal (digestion) of the gelatin film noted. The gelatin was digested by the trypsin in the stool in more than 95% of non-CF infants whereas 209 of 220 (95%) of stool specimens from untreated infants with CF showed no tryptic activity whereas less than 5% of 500 controls lacked tryptic activity. Three separate stool specimens were recommended for diagnosis.
The photograph in the paper (Fig.17) shows gelatin film acted upon by stool suspensions from three individuals (1, 2 and 3). Concentrations of 1/5 on upper row and 1/10 are on the lower row being the more dilute.
1- Normal infant’s stool with extensive white areas where there has been extensive digestion of the gelatin by the trypsin in the stool.
2- Infant with cystic fibrosis – no digestion of gelatin.
3- CF infant on treatment with pancreatin now digesting gelatin.
Subsequent papers on the gelatin film tests discussed problems with interference from faecal bacteria digesting the gelatin on the film (e.g. Johnstone DE, Neter E. Pediatrics 1951; 7:483-490). Bodian (1952) notes that Dorothy Andersen was the first to suggest testing for stool trypsin – an extremely important test prior to the availability of the sweat test for there were difficulties in duodenal intubation in small infants to obtain fluid for trypsin estimation.
– Before the sweat test became available during the Fifties, demonstration of a pancreatic enzyme abnormality was mandatory in making a correct diagnosis of CF (which was essentially a death sentence for the infant) and differentiating the condition from coeliac disease from which the infant may recover. Duodenal intubation to measure tryptic activity was the method recommended by Dorothy Andersen.
1949 Andersen DH. Therapy and prognosis of fibrocystic disease of the pancreas. Pediatrics 1949; 3:406-417. [PubMed]
Andersen still maintained that the respiratory infection was the result of nutritional failure including malabsorption of vitamin A and other fat-soluble substances. There is considerable discussion on the merits of the nutritional and the congenital hypotheses – re. the latter it was noted that “there is no morphologic evidence that a specific congenital anomaly exists in the bronchi or their secretions”. There seemed to be reluctance to finally abandon the vitamin A role in the causation of the secretory abnormalities.
1949 Andersen DH. The present diagnosis and treatment of cystic fibrosis. Proc R Soc Med 1949; 42:25-32. [PubMed]
Dorothy Andersen gave a lecture at the Royal Society of Medicine in London. She considered three groups of patients – those dying of intestinal obstruction at birth, those with onset of chronic cough before 6 months and those with onset of cough after 6 months. Andersen noted that “the lung changes were formerly believed to result from vitamin A deficiency but recently it has been suggested that they too are the result of abnormal bronchial secretions”. It is interesting that elsewhere in the same year 1949 (Pediatrics 1949; 3:406-417. above) Andersen still maintained her oft-stated belief that “persistent bronchitis complicating fibrocystic disease is the result of failure of absorption of vitamin A and presumably other fat-soluble specific substances which in turn results from an inability of the diseased pancreas to secrete enzymes necessary for absorption of fat.”
1949 Harper MH. Congenital steatorrhoea due to a defect of the pancreas. Med J Australia 1949; 1:137-141. [PubMed]
Having reported 2 cases in 1930 and 8 more in 1938, (both reviewed above) Margaret Harper from Sydney now reports 42 patients with steatorrhoea secondary to pancreatic abnormalities.
The present article is full of good old-fashioned clinical observations e.g. the stools “greasy, pale yellowish green appearance and oil may be passed with the stool or separately”. Margaret Harper states that “One condition indispensable in making the diagnosis of congenital pancreatic steatorrhoea is a personal examination of the stools interpreted in the light of the clinical examination of the child”. “Butter stools were noted in 25 cases”. As a practicing paediatrician herself, who had published two important papers on CF in 1930 and 1938 (both above) Margaret Harper draws attention to the fact that Dorothy Andersen was a pathologist and writes “Andersen’s article from the pathological laboratory of the Babies Hospital New York reported 49 cases culled from various sources; it is written from the pathological standpoint without personal clinical experience of the disease”!
Of course it is true, at the time Andersen’s 1938 paper was published her experience was based on pathology experience. Margaret Harper was obviously a paediatric clinician of the old school who was not too keen on an American pathologist (Dorothy Andersen) describing the clinical features of “so-called cystic fibrosis” from her laboratory. After all, Margaret Harper had described two such children in 1932, some 6 years before the Andersen 1938 paper, and a further 8 cases in 1938. However, although some Australians have attributed the first description of CF to Margaret Harper, it must be stated that it was undoubtedly only Andersen’s 1938 paper that resulted in a general recognition of CF as a specific entity. (More detail on Margaret Harper included in the Harper 1938 entry above).
1949 Zuelzer WW, Newton WA. The pathogenesis of fibrocystic disease of the pancreas: a study of 36 cases with special reference to pulmonary lesions. Pediatrics NY 1949; 4:53-69.[PubMed]
Autopsy details of 36 children with CF are described from Detroit. In contrast to Andersen’s belief, they found no evidence for a primary nutritional factor in the genesis of the pulmonary changes. Squamous cell metaplasia previously considered to be related to vitamin A deficiency, was found in the trachea and bronchi in only 15 of the 28 cases in the respiratory group and was never present outside the respiratory tract. “The plugging of the bronchi and bronchioles with secretions was the earliest change observed and was invariably associated with emphysema and atelectasis. The bronchial walls remained intact, inflammatory changes were as yet absent and no squamous cell metaplasia of the respiratory epithelium was found.” These findings tended to support the theory developed by Farber of the basic pathologic process being an anomaly of secretory function involving a number of organs – respiratory tract, intestine and biliary system. Nutritional deficiencies were considered to play only a secondary, though clinically very important, role. Incidentally three black children were included in this series.
The authors state -”Our findings seem conclusive evidence that the involvement of the respiratory tract is an integral part of the disease which develops parallel with, not secondary to, the nutritional failure and depends on a basic abnormality of secretory processes predisposing to secondary infection” – a clear repudiation of Andersen’s vitamin A theory of pathogenesis and, as it turned out, an accurate prediction.

Fig.18. Richard Boucher
Comment by Dr Richard Boucher (Fig.18), of Chapel Hill North Carolina, who is the champion of the low salt theory of pathogenesis. Low chloride secretion and excessive sodium absorption due to defective CFTR, in the airway epithelial cells, results in reduced airway surface liquid, interfering with removal of bacteria and other foreign substances – hence the predisposition to infection and difficulty clearing infected material. Speaking at the Royal Brompton Hospital in London in 2007, Richard Boucher said he regards this as his favourite paper on cystic fibrosis in the literature – so a few words about the first author, Dr Zuelzer, and his interesting and varied career!

Wolf W Zuelzer
Wolf William Zuelzer (Fig.19.) (1909-1987) was born in Berlin to Edith and Georg Ludwig Zuelzer-Wolff (1870-1949) His father was physician, professor in Berlin and himself son of the physician Professor Wilhelm Zuelzer (1834-1893). Wolf studied French literature at Sorbonne 1927-28, then 1929-30 philosophy and Romance languages at the University of Heidelberg with Karl Jaspers (1883-1969) and Ernst Robert Curtius (1886-1956) as teachers. After further studies in Paris and Germany, within 6 months of completing a PhD, he abruptly switched to medicine in 1930.
He studied medicine at Bonn and Berlin until he was expelled from this university in 1933. He achieved his studies at the German University of Prague, where he earned his M.D. degree in 1935. Then he immigrated to the United States. After a paediatric internship at the Massachusetts General Hospital and a year as an unpaid volunteer in pathology at the Boston Children’s Hospital under Dr. Sidney Farber he went in 1941 to Detroit. At the Chicago Children’s Hospital he was trained in pathology and paediatrics under Dr. Joseph Brenneman. In 1945 he was appointed as assistant professor of pathology and paediatrics, in 1947 as professor of pediatric research. In 1949 he received the Mead Johnson Award from the American Academy of Pediatrics for discovery of megaloblastic anaemia of childhood. From 1955 to 1975 he was director of the Child Research Center and Medical director of the Michigan community Blood Center. From 1976 to 1979 he headed the division of Blood Diseases and Resources of the National Heart, Lung and Blood Institute, Bethesda, Maryland. In 1976 he received the Grove Rasmussen Award of the American Association of Blood Banks. (from Whonamedit? A dictionary of medical eponyms).(Audrey K Brown. Dr Wolf W Zeulzer – A unique Phenotype Presentation of the Howland Award. Pediatric Research 1985; 19 (12):1365-1368).
1949 Pugsley HE, Spence PM. Case of cystic fibrosis of pancreas associated with chronic pulmonary disease and cirrhosis of the liver. Ann Int Med 1949; 30:1262-1272. [PubMed]
Cirrhosis of the liver in 17 year old white boy with CF and chronic pulmonary disease. At post mortem the liver was grossly nodular and microscopically the portal areas showed extensive irregular fibrosis with proliferation of the small bile ducts.
1949 Matheson WJ. Fibrocystic disease of the pancreas. BMJ 1949; 2(4620):2006-210.[PubMed] (Free PMC article available)
William James Matheson (1915-1993) eventually was consultant paediatrician in Leicester but working at St Mary’s at this time. He notes that there are few reports from the UK of children with CF although the American literature was by this time fairly extensive. The report formed the basis of an MD thesis and the children were under the care of a distinguished paediatrician, Dr Reginald Lightfoot, at St Mary’s Hospital, London.
This is a detailed report of seven children, five of whom had died and a review of the literature to that time. They represented 2% of admissions to the children’s ward. Surprisingly, inheritance by incomplete dominance is postulated although Andersen and Hodges (1946 above) had already shown the condition to be recessively inherited. The author notes that pathological findings confirm this is a disease of mucus secreting glands throughout the body. The diagnosis can usually be made clinically and the condition differentiated from coeliac disease (according to Sir Wilfred Sheldon – a recognised expert on coeliac disease) and treatment is “disappointing”.
Please note The full version of the paper is available through PubMed and contains illustrations of a patient and some very clear histology figures showing typical findings in the lungs and pancreas. The description of the clinical course of the patients gives an idea of the management of children with CF at that time in the UK and their appalling outlook..