The Thirties


Thirties
Pancreatic malabsorption as distinct from coeliac disease – “fibrocystic disease of the pancreas” is recognised
The major advance towards the end of the Thirties was the clear description of “fibrocystic disease of the pancreas” by Dorothy H Andersen, from the Pathological Laboratory, Babies Hospital and the Department of Pathology, Royal College of Physicians and Surgeons, Columbia University, New York in a paper she read at a joint meeting of the American Pediatric Society with the Society for Pediatric Research on May 5th 1938 (Andersen, 1938 below).
Prior to the Thirties there had been isolated case reports of children with the clinical features of intestinal malabsorption considered to be due to pancreatic insufficiency (described in ‘Early Years’ section above). In some cases this suggestion was supported by the presence of pancreatic histological changes at autopsy – a feature that eventually led to the recognition of cystic fibrosis as a distinct clinical entity. Many of the children also had severe respiratory problems. Many reports contained autopsy details as most affected children died in infancy for there were no antibiotics available until the mid-Forties; even sulphonamides, which were of limited value in CF, were not available until the end of the decade. A further problem was that death in infancy from pneumonia or gastroenteritis, even in non-CF infants, in the Thirties was a relatively frequent occurrence.
During the Thirties further small series of children were reported who had intestinal malabsorption with evidence of pancreatic abnormalities. A number of authors noted that children with coeliac disease were not uniform and some had definite pancreatic abnormalities – usually noted at autopsy (Parmalee, 1935 below; Hess & Saphire, 1935 below). In 1952 Martin Bodian, pathologist at the Hospital for Sick Children, Great Ormond Street, London, commented that Dorothy Andersen’s classic 1938 paper (Andersen, 1938 below) was, in part, the result of her gradually collecting details of her own patients with pancreatic abnormalities and similar patients published by previous authors or made available by colleagues. With this information she was able to build up a picture of a specific condition in which a pancreatic lesion was often associated with steatorrhoea and respiratory disturbances (Bodian, 1952 below). During the Thirties other workers were reaching the same conclusions as Andersen at much the same time; these included in 1938 Blackfan and May (1938 below) who reported 35 cases of fibrocystic disease from among 2500 paediatric autopsies; these authors gave credit to Kenneth Wolbach to whom they attributed the first description of the typical pancreatic lesion and for the suggestion that the changes may be due to an abnormal secretion within the pancreatic ducts (Blackfan & Wolbach, 1933 below). There were others who recognised the clinical features of CF as distinct from coeliac disease (Parmalee, 1935; Hess & Saphire, 1935; Harper, 1938; Rauch et al, 1939 – all their publications are reviewed below). Although the first clear description of CF has been attributed to Fanconi (Fanconi et al, 1936 below) by some European clinicians, he described only two patients and the paper did not lead to a general recognition of the condition as did that of Dorothy Andersen in 1938.
At the end of the Twenties there had been an observation made by Alexander Flemming that eventually led to one of the most important medical advances of all time – an advance, that when eventually developed by others, was to have a major influence on the survival of children with cystic fibrosis. In 1929 Alexander Fleming, the microbiologist at St Mary’s Hospital in London, observed inhibition of a growth of Staphylococcus aureus on a culture plate by a Penicillin mould which had contaminated the plate (Flemming, 1929 above). Flemming did little more with his discovery at the time but at the end of the Thirties this initial observation eventually led to the development of penicillin by Howard Walter Florey and his colleagues in Oxford. In 1940 a pure extract of penicillin was produced from the mould Penicillium notatum by the chemist Ernest Boris Chain. Until the availability of penicillin and subsequently other antibiotics, infants with CF rarely survived beyond early childhood.
Most of the following papers describe children who are likely to have had cystic fibrosis.
Papers published in the Thirties
Please note the papers mentioned in this section are not included in the “Topics” section.
1930 Harper M. Two cases of congenital pancreatic steatorrhoea with infantilism. M J Aust 1930; 2:663-664.

Fig 1. Margaret Harper
Margaret Harper (1879-1964) was a distinguished paediatrician at the Royal Alexandra Hospital for Children, Sydney. She reported two children with typical clinical features and stools characteristic of pancreatic malabsorption; both children died of bronchopneumonia and the one autopsied had typical pancreatic changes of cystic fibrosis. Harper reported 8 further children in 1938 and 42 more in 1948. Both are reviewed below with further career details and images of Dr Margaret Harper (1938 Harper entry below).
1931 Leipmann H, Hoffman H. Investigation of a case of pancreatic insufficiency. Zeit Kinderheilk 1931; 50:212-223. (German).
A report of a child with evidence of intestinal malabsorption, laboratory evidence of pancreatic insufficiency and also keratomalacia. There was improvement with nutritional therapy but no evident benefit from Pankreon (a pancreatic extract). The passage of oil from the anus was a feature – a very typical sign of pancreatic steatorrhoea. Although there was marked steatorrhoea there was adequate carbohydrate absorption – also a feature of CF. The authors reviewed previously reported cases of pancreatic insufficiency in childhood and noted that the condition was rare.
– Almost certainly this infant had cystic fibrosis. The passage of yellow-orange oil with the stools is very suggestive, in fact virtually diagnostic, of pancreatic steatorrhoea.
1931 Daffinee RW, Higgins HL, Mallory TB. Corneal ulcers and roughened conjunctivae associated with fat intolerance. N Engl J Med 1931; 204:1264-1267.
A white male infant aged six months had spots on his eyes noted by the mother also weight loss. He died of bronchopneumonia one month later and at one stage was excreting 40% of his dietary fat intake (70% of which was neutral fat). Autopsy showed almost complete atresia of the cystic duct and histologically “extremely marked disease of the pancreas” – practically all the acinar tissue had disappeared. The lungs showed multiple abscesses. Mallory, the author who was the pathologist, writes “in the later stages of the disease he had a definite pancreatic lack whatever the original cause may have been”.
– There can be little doubt that this infant had CF although there are a number of reports suggesting that severe stenosis or even atresia of the pancreatic duct may be the primary cause of the clinical syndrome.
Ralph Weir Daffinee (1903-1959) was an instructor in paediatrics at Harvard and Tufts Medical School for 25 years
1932 Parsons LG. Celiac disease. Rachford Memorial Lectures. Am J Dis Child 1932; 43:1293-1346.

Fig 2. Sir Leonard Parsons
Sir Leonard Parsons (1879-1950) (figure 2) was Professor of Paediatrics in Birmingham, UK. Born and educated in the Midlands, Sir Leonard Parsons made major contributions to the field of paediatrics in that area and played a leading role in the regional organisation of this specialty throughout the United Kingdom in the later years of his life. He was a founder member and later President of the British Paediatric Association, and later became Vice-President of the International Pediatric Congress and the President of the paediatric section of the Royal Society of Medicine (Dunn PM. Arch Dis Child Fetal Neonatal Ed 2002;86:F65-F67).[PubMed]
This is a long article describing the current situation regarding coeliac disease drawing on the author’s experience of 73 children – 21 were from the Boston Children’s Hospital “through the kindness of Dr Kenneth Blackfan”. Parsons advised against diagnosing coeliac disease unless there were more than 2 g of fat daily in the faeces – usually there was considerably more – up to 20 g; also the percentage of fat in the stools may reach 80% – the normal upper limit being 25%. There is a detailed clinical commentary with attention to the nutritional deficiencies.
The mortality of only 10.6%, in the pre-antibiotic era, suggests that there were few infants with CF included in this series. In discussing the morbid anatomy Parsons mentions that “in two or three cases some excess of fibrous tissue or small-celled infiltration around the ducts of the pancreas has been found”. However, most authors had not found pancreatic changes at autopsy in children considered to have coeliac disease. “In celiac disease the pancreatic juice is normal because the fats in the stool are split normally; furthermore, specimens obtained by duodenal intubation have shown the presence of trypsin, amylopsin and lipase in normal amounts – when estimated by the method of McClure, Wetmore and Reynolds”. However, Parsons observed that Fanconi apparently believed “that the diastatic ferments function poorly” in coeliac disease.
So in this very long detailed paper by one of the UK’s leading paediatricians, most of the discussion centered on what was almost certainly gluten induced coeliac disease; the possibility that some cases were due to pancreatic abnormality was discussed only briefly but dismissed.
Many years later some degree of pancreatic insufficiency was reported to occur in untreated coeliac disease but this was only temporary and considered to be due to substrate protein deficiency associated with the malnutrition (Carroccio A et al, Gut 1991; 32:796-799. [PubMed]).
1932 Van Creveld S. Pancreatic fat stool of children. Ned Tidjschr Geneeskd 1932; 76:3741-3755. (German)

Simon van Creveld Het NVK archief
“In describing some morbid cases the question is extensively discussed in how far fat stool in children may be caused by a primary pancreatic affection. Especially the differential diagnosis between intestinal infantilism and pancreatic fat stool is stressed; a case of the latter condition is described.”
Simon van Creveld (1894-1971) was appointed professor of paediatrics in Amsterdam in 1933 but removed by the Nazis in 1941. He was in a concentration camp along with his wife during the war and re-instated as professor after the war. “During the Nazi occupation of the Netherlands, van Creveld’s professorship was taken away from him because he was Jewish. His visits to hospitals of concentration camps to treat babies and give pediatric advice while wearing a Jewish Yellow Star and interacting with SS Commandants in charge, and then leaving can only be described as amazing. After the war, his professorship was returned, and in the same year as his retirement, he established a large Hemophila Treatment and Research Center now known as the Van Creveld Clinic, which celebrated its 40th anniversary in 2005” (extract from Paul JW Stoelinga et al. pubmed.ncbi.nlm.nih.gov/25269937/).
1932 Siwe S. On the exocrine function of the pancreas and the results of their omission. A case of almost total agenesis of the exocrine portion of the pancreas. Deutsche Arch Klin Med 1932; 173:339-358. (German).
A child with feeding problems from the age of six months and cough from 19 months died aged two years. She had been born prematurely and had foul stools, poor development and a large abdomen. She died from varicella and bronchitis. The pancreas had well-preserved islets in contrast to almost total degeneration of the exocrine portion of the gland. There was rickets and mild cirrhosis and a fatty liver (details from Andersen, 1938. below).
This patient was accepted by Dorothy Andersen (Andersen, 1938) as having CF, although rickets is rare in cystic fibrosis; however, it is more likely to occur if there is liver disease (Scott J et al, 1977. below).
1933 Blackfan KD, Wolbach SB. Vitamin A deficiency in infants. J Pediatr 1933; 3:679-706.

Fig 3. Kenneth Blackfan
Kenneth Blackfan (1883-1941) (figure 3) was Associate Professor of Pediatrics at Johns Hopkins, then Professor of Pediatrics at Cincinnati and at Harvard from 1923 to 1941. He apparently ‘mentored’ both Louis Diamond and Sydney Farber. In 1941 he died of lung cancer at the age of 58 – at the height of his career. The Children’s’ Hospital in Boston is on Blackfan Street – named after him.
An interesting detailed account of Kenneth’s Blackfan’s very distinguished and varied career was published in 2015. (Jeelani, Y., Cohen, A.R. The Gentle Giant: Kenneth Daniel Blackfan and his contributions to pediatric neurosurgery. Childs Nerv Syst 31, 821–831 (2015). https://doi.org/10.1007/s00381-015-2658-x).
This paper contains an interesting early account primarily of vitamin A deficiency, in which there was considerable interest at the time – the vitamin having been discovered in 1917. In 13 infants and young children, 11 of whom were eventually autopsied, vitamin A deficiency was reported. Epithelial metaplasia due to vitamin A deficiency was considered an important predisposing factor to respiratory infection by causing – “loss of protective powers of the epithelium due to diminished or absent mucus secretion and loss of ciliary motion”. Six of the 11 infants autopsied had extensive pancreatic lesions (later recognised as typical of CF), that the authors correctly attributed to inspissation of secretion in the ducts.
The authors noted “The pancreatic lesions were all identical and presumably representing a disease entity. At first we regarded the pancreatic changes as the result of vitamin A deficiency (as did Dorothy Andersen for some years). As the same condition has been found scores of times without other evidences of vitamin A deficiency and since it is not constant even in vitamin A deficiency, we must consider the two are not necessarily connected”. The photomicrograph of pancreas (figure 4a) is typical of the changes seen in CF for which Blackfan & May (1938 below) gave credit to Wolbach for the first clear histological description.
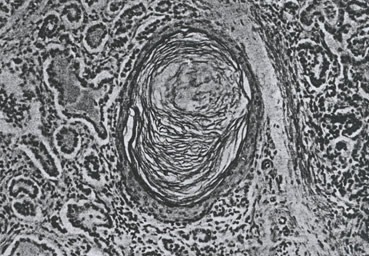
Fig. 4a: Photomicrograph of pancreas. From Blackfan & Wolbach, 1933 with permission.
The authors continue “The pancreatic lesion referred to is characterized by dilatation of acini and ducts, by inspissated secretion, atrophy of the acini, lymphoid and leukocyte infiltration of some degree and fibrosis. Our preliminary studies indicate that the pathogenesis of this striking pancreatic affection resides in the production of an abnormal secretion which inspissated and leads to distension and atrophy of ducts and acini. It is reasonable to assume that this pancreatic lesion, if extensive, may be responsible for failure to utilise fats and hence vitamin A in the presence of an adequate intake”.
The authors concluded that vitamin A deficiency was not infrequent and should be suspected without regard to the characteristic eye signs; histological evidence of vitamin A deficiency may appear in infants who appear to be receiving and adequate intake; they postulate that some factor or factors interfering with storage or utilisation of this vitamin lessens the availability of the supply essential for normal nutrition; certain diagnostic criteria to identify at an early stage are mentioned and vitamin A deficiency should be considered as a general systemic disease rather than as a local disease of the eye.

Fig 4. Simeon Wolbach waywiser.fas.harvard.edu
Dr Simeon Burt Wolbach (figure 4) (1880-1954) was born in 1880 in Grand Island, Nebraska, and grew up on the western plains. He had a veried and interesting career. He attended Harvard Medical School (M.D. 1903), studying pathology under Councilman and Mallory. After medical school, he worked as director of the Bender Hygienic Laboratory in Albany, NY; pathologist at the Montreal General Hospital and teacher at McGill Medical School. In 1910, he returned to Harvard as an assistant professor of bacteriology. He was promoted to associate professor in 1914. In 1916, he was jointly appointed to the Departments of Bacteriology and Pathology. In 1917, he was pathologist to the Peter Bent Brigham Hospital and the Boston Lying-In Hospital. In 1922, he was appointed the Shattuck professor of pathology at Harvard. He held this position for 25 years. In 1947, he became the director of the Division of Nutritional Research at Children’s Hospital in Boston. He held this position until his death in 1954.
Wolbach’s interests ranged from the effects of radiation to tropical medicine and infectious diseases. His work on radiation began with Porter in 1907. Later in life, he served as a consultant to the US Atomic Energy Commission (1951-1953). He did field research in Nigeria in 1911 related to the pathology of general paresis. Best known of his work on infectious diseases are his contributions to the understanding rickettsial illnesses, Rocky Mountain spotted fever (1919), and epidemic typhus in Poland (1920). However, Wolbach’s most fundamental work was in the field of vitamin research, and the relationships of vitamins to tissue structure and the pathology of scurvy and other diseases.
1933 Greenberg J. An attempt to reproduce coeliac disease experimentally in young animals by excluding the external pancreatic secretion from the intestine. Yale J Biol Med 1933-34; 6:121-153.
In this paper there is a previously unreported case described of an infant aged 11 months in whom a completely fibrotic pancreas was found. The very title of this paper suggests that the differentiation between coeliac disease and CF was still far from clear.
1934 Huet GJ. A case of Herter’s infantilism with chronic lung involvement. Maandschr v Kindergeneesk 1933-34; 3:343-350. (Dutch).
A boy aged 10 years who had chronic diarrhoea died of bronchopneumonia. There had been feeding difficulty in the infancy and a chronic cough from the age of six years. His development was generally poor, his stools fatty and he had chronic chest infection. Autopsy showed purulent bronchitis with dilation of the bronchi and fibrosis of the lungs. There was marked atrophy of pancreatic acini and hypertrophy of the islets; also fatty degeneration and cirrhosis of the liver. Tuberculosis had been suspected during life but was ruled out.
This patient is mentioned by Andersen (1938 below) and almost certainly had cystic fibrosis.
1935 Anke H. A case of congenital pancreatic insufficiency. Munchen Med Wochen 1935; 82:1787-1789.
A boy aged 14 years with clinical evidence of congenital pancreatic malabsorption with steatorrhoea; he had typical “butter stools” containing an excess of neutral fat. There were no significant chest problems – he even did well in the Hitler Youth movement!
The overall progress, normal growth, lack of respiratory problems are against this boy having CF although he could have had atypical CF due to unusual so-called “mild” mutations. However, the “butter stools” would suggest pancreatic malabsorption and could indicate that he had “congenital lipase deficiency”, later described by Sir Wilfred Sheldon (1901-1983), paediatrician at The Hospital for Sick Children, Great Ormond Street, London (Sheldon W. Arch Dis Child 1964; 39:268-271. [PubMed]; also similar by Ligumsky M et al. Gut 1990; 31:1416-1418). [PubMed]. Both of Sheldon’s children had severe steatorrhoea, a tendency for oil to ooze from the anus, but satisfactory growth, absent or low pancreatic lipase but normal amylase and proteolytic enzymes in the pancreatic juice.
A similar patient was described by Garrod & Hurtley (1912 above).
1935 Parmelee AH. The pathology of steatorrhoea. Am J Dis Child 1935; 50:1418 -1428.

Fig 5. Arthur Parmelee
Parmelee (figure 5) described a girl who died aged 14 years. She had early feeding problems, fat intolerance with rancid fatty stools, a protuberant abdomen, bronchitis and eventually bronchopneumonia; at autopsy her pancreas showed the typical changes of cystic fibrosis. Another girl who died aged four years had poor weight gain and the appearances of intestinal malabsorption. At autopsy the pancreas was typical of CF with a narrow duct which was queried as atretic. “Congenital steatorrhoea” and “pancreatic steatorrhoea” obviously referred to infants who had CF and differed from the usual picture of coeliac disease in their early onset, excessive amount of neutral fat in the stools and the uniformly fatal outcome. There were constant changes in the pancreas at autopsy – “marked fibrosis and great diminution of secretory gland tissue in the pancreas, also bronchopneumonia”.
Arthur H Parmelee (1883-1961)(figure 4a) of Chicago was a highly respected paediatrician from Chicago where he graduated at Rush Medical College in 1911. For a time he was also football coach while in Miami. After some years in general paediatric practice, in 1924 he departed for Vienna, Austria to study with Clemens von Pirquet, at the time the leading paediatrician in Europe, He would return to Vienna for additional study in 1931 and 1932. In 1947 he left his private practice, resigned from the pediatric department at Rush and moved to Los Angeles where he became a member of the Staff at Children’s Hospital Los Angeles. In addition he was a Pediatric Consultant to the Bureau of Maternal and Child Health of the California State Department of Public Health, and Clinical Professor of Pediatrics at the USC School of Medicine.
This paper is mentioned frequently in the literature as making a major contribution to clarifying the situation regarding the various conditions classified as coeliac disease by correlating the clinical and pathological picture. Charles May (1954 below) obviously considers Margaret Harper and Arthur Parmalee to be the first to describe pancreatic steatorrhoea. May writes a tribute – “To the practitioners, Margaret Harper (1938 below) of Sydney, Australia and Arthur H Parmelee of Chicago, Illinois (1935 above) who recognised the salient clinical features of patients found to have cystic fibrosis of the pancreas, published the first papers indicating the frequency and importance of the disease, and clearly set it apart from celiac disease against the prevalent practice”.
Interestingly Dorothy Andersen is not mentioned by May in this context! Also May recalls how Blackfan was steadily preoccupied during the Thirties with the problem of recognising infants who had cystic fibrosis of the pancreas culminating in his report of 1938 (Blackfan & May, 1938 below).
1935 Hess JH, Saphire O. Celiac disease: Chronic intestinal indigestion. A report of three cases with autopsy findings. J Pediatr 1935; 6:1-13.

Fig 6. Julius Hess
This is a frequently quoted paper from Chicago describing three infants with pancreatic insufficiency and severe pancreatic fibrosis at autopsy with distension of the ducts, also liver involvement and bronchopneumonia with abscess formation.
There is an interesting account of the published views on the aetiology of coeliac disease, noting that only some authors describe associated pancreatic lesions.
The present authors concluded that “If, by the use of special staining methods, it is not possible to demonstrate fibrosis in the pancreas, then it must be conceded that the symptom complex of celiac disease has not one definite pathological entity…..Because of our findings we must insist, however, that in every instance of celiac disease the pancreas should be studied with the before mentioned special methods before pronouncing it normal”.
Here was further evidence that coeliac disease was really a syndrome comprising more than one condition and good advice to always exclude pancreatic abnormality. So by the mid-Thirties cystic fibrosis was gradually emerging as a specific entity.
Dr Julius Hess (1876-1955) was an American physician often referred to as the father of American neonatology. He published a textbook on the care of pemature infants and those with birth defects in 1922 and with nurse Evelyn Lundeen created the first premature infant station in the United States. (from Dunn PM. Julius Hess MD (1876-1955) and the premature infant. Arch Dis Child Neonatal Ed 2001:85:F141-144).
1936 Dodd K. Intestinal obstruction due to meconium ileus in a newborn infant. J Pediatr 1936; 9:486-491.
Katherine Dodd of Nashville notes that “from time to time there have appeared in the literature reports of obstruction due apparently to no tumour or congenital malformation of the gastrointestinal tract but to an accumulation of mucilaginous meconium in the tract itself”. There is usually no mention of meconium ileus even in the more complete textbooks. This infant had signs of intestinal obstruction, developed bronchopneumonia and died at 10 days. There was tenacious grey material in the ileum and colon. “The only other organ showing marked changes was the pancreas which showed marked changes of interstitial pancreatitis, cellular infiltration and dilatation of some ducts and acini” – in fact it was typical of cystic fibrosis.
A review of the literature by the author revealed 21 similar reported cases – the five who recovered were well having passed a large meconium plug and they probably had “meconium plug syndrome” and not cystic fibrosis. The rest had fatal obstruction due to abnormal bowel contents. In five cases abnormalities were found in the pancreas where this was examined (Kornblith et al, 1929 above; Landsteiner, 1905 above). Katherine Dodd suggested that the failure of pancreatic secretion or bile to reach the gastrointestinal tract during fetal and neonatal life was at least part of the cause of the meconium ileus.

Fig 7. Katherine Dodd
Katherine Dodd (1895-1965) (figure 7) received her M.D. from Johns Hopkins in 1921. After nine months in Bulzulnek, USSR, assisting at a Quaker clinic for child welfare and famine relief, in 1926,she was recruited by Canby Robinson for Vanderbilt’s Department of Pediatrics, where she remained for the next eighteen years. She quickly acquired a reputation as an outstanding clinician and researcher, and was highly popular with students. However, she was eventually passed over for the Department’s chairmanship. In 1944, she left Vanderbilt to accept a position at Cincinnati’s Children’s Hospital Research Foundation. Later, Dr. Dodd served as Professor of Pediatrics at the University of Arkansas, the University of Louisville, and Emory University. In the summer of 1947, she was a member of a team sent by the US Public Health Service to Japan. The team succeeded in its mission of investigating the cause and treatment of ekiri, a highly fatal disease prevalent among Japanese children.
Her later years were marked by numerous awards and honors. On the 75th anniversary of Bryn Mawr College, Dr. Dodd was honored as one of its most outstanding alumnae. (from pediaitrics.mc.vanderbilt.edu)
1936 Fanconi G, Uehlinger E, Knauer C. Das Coeliakie-syndrom bei angeborener zystischer Pankreasfibromatose und Bronchiektasien. Wien Med Wchnschr 1936; 86:753-756. (Celiac syndrome with congenital cystic fibromatosis of the pancreas and bronchiectasis).
I am grateful to my friend and erstwhile St James’s surgical colleague Mr Archie Crompton for translating this article for me.

Fig 8. Guido Fanconi
Guido Fanconi (1892-1979) (figure 8) was Professor of Paediatrics in Zurich from 1929-1965 and a pioneer of modern scientific paediatrics. Although this publication is considered by some Europeans to be the first clear description of CF, it is a brief report of only two children who died aged 10 months and three years with ‘coeliac syndrome’, purulent bronchitis and bronchiectasis. The pancreatic changes were quite typical of those later described in cystic fibrosis. “The changes in the lungs and pancreas, two vital organs, are so profound that their failure appears understandable”.
Martin Bodian quotes a 1928 publication of Fanconi’s (1928 Beih. Jb. Kinderhlk 21) as noting cases of coeliac disease starting in early infancy and often associated with bronchitis but surprisingly Fanconi did not mention this in this 1936 paper which is so frequently quoted in Europe as the first description of cystic fibrosis.
Speaking in 2008, Walter Hitzig, the distinguished immunologist from Zurich, recalls (referring to this paper) that “My teacher Guido Fanconi, in his usual enthusiasm, presented a “new disease” to the Swiss paediatricians in 1936. However, one of the doctors questioned its importance in his daily practice. Fanconi’s answer: “Oh certainly it is important – I have seen already 3 cases!” of course earned him a big laughter in the audience! And today the syndrome described as Pankreas-Fibrose, now called cystic fibrosis, is considered to be the most frequent hereditary disease in the Caucasian population, and no doubt every paediatrician must diagnose it”.
Lynn Taussig describes meeting Knauer, the third author of this paper, after it was published. Apparently the two children described in Knauer’s doctoral thesis in 1935 were the two described in this paper and the next year Fanconi, Uehlinger and Knauer published this paper. He notes that the first name on the paper was now Fanconi’s!
Maurice Super mentions that there was controversy between Fanconi and Wangensteen “who thought the condition might be something unique to the cantons in which Fanconi worked”. The hereditary nature of CF was not suspected at this time.
Fanconi published a number of papers on various aspects of intestinal malabsorption and other aspects of paediatrics between 1921 and 1956 mostly in German or French. He was referred to in one article as a “Jack of all trades” – as were many of the paediatricians of the time. Reviewed objectively, none of these publications, with the possible exception of this 1936 paper, made any major or original contribution to the understanding or definition of cystic fibrosis. However, Fanconi made other very important contributions; in 1929 he described hereditary panmyelopathy known as Fanconi’s anaemia, in 1941 he deduced that polio was spread via the gastrointestinal route rather than by droplets and apparently predicted that Down’s syndrome was due to a chromosome abnormality 20 years before trisomy 21 was discovered
Addendum 2020. Jurg Barben. First description of cystic fibrosis. J Cyst Fibros 2021; 20(1): 183

Fig.8a. Jurg Barben
Professor Jurg Barben (Fig.8a) discussing the widespread cited article by Dorothy Andersen in 1938 as the first description of cystic fibrosis, draws attention to the fact that Guido Fanconi’s described two children in 1936 who almost certainly had cystic fibrosis as judged by the autopsy findings. In addition to Andersen’s substantial numbers, her article was in English and Fanconi’s was written in German and published in a German language journal (The Vienna Medical Weekly). Barben observes this publication has now been forgotten in the English-speaking world. However, a German-American colleague, Dr Teodore Bruns (former CF Centre Director in Milwaukee, USA) did a personal translation of Fanconi’s publication many years ago which was never published. This English translation in now (with Permission of Phillip Farrell, Emeritus Dean and Professor for Pediatrics and Population Health Services at the University of Wisconsin, USA who provided the translation) available on line in order to preserve it for the next generation of researchers. https://www.kispisg.ch/downloads/kompetenzen/pneumologie/fanconi-1936_english-translation.pdf
1936 Meltzer S. Meconium ileus. Canad Med Ass J 1936; 34:186.
A single case report by Sara Meltzer, a pathologist in Winnipeg – the second infant in the family to die of neonatal obstruction. Abnormal meconium was noted but pancreas was not examined and the infant died on the first day. She mentions a similar case report by Kornblith & Otani 1929 (above) where there was stenosis of the duct of Wirsung (the main pancreatic duct) also twins seen by Sydney Farber with occlusion of the pancreatic duct and pancreatic fibrosis (also Dodd, 1936 above).
Dr Sara Meltzer (1900-1942) joined the staff of the Winnipeg General Hospital in 1924 where she was assistant pathologist.
1936 Garsche R. Clinical forms and pathogenesis of pancreatic insufficiency in older children. Z Kinderheilk 1936; 58:434-455. (German).
An infant with feeding problems from 6 months who died aged four years and six months after developing chest problems. He had failure to gain weight, with large foul stools, protuberant abdomen, and alternate hunger and anorexia. The cause of death was pneumonia. The pancreas showed fibrosis with replacement of acinar tissue with fat and concretments. The liver was fatty with mild biliary cirrhosis (mentioned by Andersen as having CF, 1938 below).
1938 Burger P. Cas rare d’ileus du nouveau-ne par epaississement du meconium. (A rare case of ileus in the newborn with inspissation of meconium). Gynec et Obstet 1938; 37:176-177.
A case of neonatal intestinal obstruction with pancreatic changes at autopsy. Almost certainly meconium ileus attributed to some abnormality in the secretions of the liver or pancreas.
1938 Thomas J, Schultz FW. Pancreatic steatorrhoea. Am J Dis Child 1938; 56:336-343.
This study from Chicago identified 10 previous reported cases of pancreatic steatorrhoea (Garrod & Hurtley, 1913; Miller & Perkins, 1920; Clarke & Hadfield, 1924; Harper, 1930; Siwe, 1932, Hess & Saphir, 1935; Parmalee, 1935 – all reviewed above).
The authors report an emaciated boy who died aged five years with progressive chest infection and steatorrhoea. There was typical CF histology of an atrophic and markedly fibrosed pancreas.
There is a footnote that Margaret Harper had published a report of 8 more cases since this present paper was submitted (Harper 1938, below)
Further authors collecting examples of infants who had similar clinical and histological features and suggesting they had a specific condition.
The following three papers were regarded by di Sant’Agnese and Andersen in 1959 as simultaneously describing cystic fibrosis although, Dorothy Andersen’s 1938 paper is usually taken as the first clear description of cystic fibrosis that prompted others to search for and recognise infants with CF – usually by reviewing their previous paediatric autopsies of which there were many in those days.
1938 Harper MH. Congenital steatorrhoea due to pancreatic defect. Arch Dis Child 1938; 13:45-56.
Further to her 1930 report of two children with steatorrhoea (Harper, 1930 above), Margaret Harper of Sydney reported a further eight children with features compatible with cystic fibrosis. Congenital steatorrhoea, abnormal glucose tolerance curves and characteristic pancreatic lesions in all the 4 who were autopsied; 8 of the 10 who died had bronchopneumonia.
“The passage of fatty stools from birth (particularly “butter stools”) with failure of nutrition, the diagnosis of congenital defect of the pancreas can reasonably be made”.
Subsequently Harper reported 42 children with pancreatic steatorrhoea from the Royal Alexandra Hospital for Children, Sydney (Harper,1949 below). There are interesting comments on Andersen’s paper in this 1949 paper (below).
The orange-yellow oil in, on and around the stools is now recognised as a typical and frequent feature of untreated cystic fibrosis. The discussion in this paper reviews previous reported cases with particular emphasis on the characteristics of the stools. A visit to the ward sluice to inspect the stools of children on the ward was a usual part of the paediatric ward round even in the Sixties and considered to be an essential part of the full evaluation of an ill child just as was chemical testing and microscopy of the urine by the ward doctor.
I am grateful to Professor John Walker-Smith for further information on Margaret Harper. He notes that she was a

Fig 9. Margaret Harper
pioneer of women in medicine and reported to have been the first lecturer in Diseases of the Newborn to be appointed in the British Empire. She was appointed as a full time paediatrician in 1914 at the Royal Alexandra Hospital for Children in Sydney and thus may be one of the first full time paediatricians.
Writing in 1979, Dr D G Hamilton described Margaret Harper (Figs 9) as – “the brilliant outstanding mind at the Royal Alexandra Hospital for Children. She continued her interest in babies and their welfare throughout her whole life. She greatly influenced the care of children with gastroenteritis with feeding problems and with chronic diarrhoea. These latter were grouped under the title of the Coeliac Syndrome. Last century Samuel Gee in Britain had talked of offensive bread when discussing these children. Margaret Harper by patient trial of diets always seemed to achieve better results with her coeliac patients than any other physician and one of the things she did was to avoid giving them cereals. They were fed copious amounts of banana and cottage cheese. In 1945 it was recognised in Holland that one of the more common causes of chronic diarrhoea in little children is intolerance to gluten that is found in wheat, rye, barley and oats and the name coeliac disease was given to this particular condition. Margaret Harper’s diet of bananas, cottage cheese and no cereal was close to the mark. She was a very careful and thoughtful observer.

Fig. 9a Frank Tidswell dictionary of sydney.org
Among these babies she recognised that a small group were different from the others. They were more prone to chest infections and their stools looked to her to be different and more greasy. These children were prone to die. She drew the attention of Dr Frank Tidswell (1867-1941) (Fig 9a) the director of pathology, to her belief that they were different from the others and at post-mortem he found characteristic changes in the pancreas. She was the first person in the world to recognise this as a separate condition and in 1930 published a report in the Medical Journal of Australia entitled “Two cases of congenital pancreatic steatorrhoea with infantilism”. Similar reports followed in 1935 by Arthur Parmalee in Chicago. In 1938 Margaret Harper published a more detailed paper on the subject in the British Archives of Diseases of Childhood entitled Congenital steatorrhoea due to Pancreatic defect. In 1954 Dr Charles May, an American expert, wrote a monograph on Cystic Fibrosis of the Pancreas (see May 1954 below) and dedicated the work to “the practitioners Margaret Harper of Sydney, Australia and Arthur H Parmalee of Chicago, Illinois who recognised…..and published the first papers, indicating the frequency and importance of the disease, clearly setting it apart from Coeliac disease, against prevalence practice. Margaret Harper was always a much loved figure at the hospital, tall, very helpful to those who tried hard, always prepared to be critical, but tempering her criticism with a laugh. She has splendid scientific mind. She entered happily and freely into bedside discussions and informal argument at clinical meetings where she was usually extremely stimulating, but she hated formal lectures. These she simply read from a prepared script in a monotonous tone and with her head down. She really was a dreadful lecturer. But she was perhaps the greatest physician the hospital has known” (DG Hamilton. Hand in Hand. The Story of the Royal Alexandra Hospital for Children, Sydney. John Ferguson, Sydney. 134-136:1979).
Much information and images of Margaret Harper kindly provided by Professor John Walker-Smith in 2010
1938 Andersen DH, Cystic fibrosis of the pancreas and its relation to celiac disease: a clinical and pathological study. Am J Dis Child 1938; 56:344-399.
Although many infants had already been reported who, in hindsight, obviously had cystic fibrosis, this was the first clear detailed clinical and pathological description of a large series of affected infants in English. Dorothy Andersen reported 49 patients – 20 from her hospital and others from colleagues and the literature. She described the neonatal intestinal obstruction, intestinal and respiratory complications and many other features – but particularly the characteristic pancreatic histology (figures 7 and 8). Salient points from the original description are as follows – “In 45 of the cases the pancreas presented a microscopic picture which is described by the term cystic fibrosis. The acini contain secretions of various sizes, and the acinar cells were flattened to form a thin epithelial wall around them. The smaller concretions were surrounded by relatively normal cells, which occasionally contained eosinophil granules. The size of the cysts varied in each case but large ones were not often noted in the youngest infants. Surrounding the acini and also the lobules there were moderate to large amounts of fibrous tissue, the quantity varying roughly with the age of the child…….The islets of Langerhans were usually normal in number and appearance.”
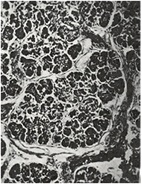
Fig. 10: Pancreas of non-CF newborn aged 3 days. With permission.
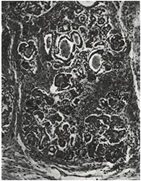
Fig. 11: Pancreas of CF newborn aged 6 days. With permission.
Andersen likened much of the epithelial histology to that found in vitamin A deficiency which for some years she regarded as the primary cause of the systemic features of the condition; this causation was never substantiated although she continued to support this theory for some years.
The paper contains an excellent tabulated review of the previously reported cases. Describing her paper Martin Bodian (1952 below) writes – “such a clear account of the symptoms that it enabled many cases to be recognised that had hitherto been missed, and aroused such interest that it was followed by a shoal of case reports and confirmatory reviews” which appeared during the Forties (many are described below).
Dorothy Andersen (1901-1963) (figure 9) was the pathologist at the Babies Hospital at the Columbia Presbyterian Medical Center in New York. She had a wide range of interests and made contributions in many other areas of medicine. She was said to be a rugged individualist, a paediatric clinician, pathologist, a research chemist – also apparently a roofer and carpenter, happy to make her own home improvements! Eventually over 600 children with CF were referred to her in New York; she involved Dr. Paul di Sant’Agnese, eventually another leader in the CF field, to provide paediatric care for her patients as she was primarily a pathologist.
Dr Phillip Farrell of Wisconsin lists 4 reasons for the referral of so many children to her in New York. First was this seminal 1938 publication, second a combination of her reputation and her arrogance (she is alleged to have said she was the only person in the world who knew about cystic fibrosis in the late Thirties and Forties!), third the desperation of parents and fourth New York was the one place people could reach easily in those days. Described as “windblown” by friends and detractors alike, Dorothy Andersen was considered quite a character. She is said to have kept a particularly untidy lab, holding semi-annual “glüg” parties there, in honour of her Scandinavian heritage. She was a niece of Hans Christian Andersen. She was a bright compassionate and sensitive scientist who was also a chain smoker and died of lung cancer at the age of 62 years.

Fig. 12: Dorothy Andersen From Wikipedia
In 1950 Milton Graub, a paediatrician who eventually became President of the US CF Foundation, suspected his two year old son had CF and describes how he called Dr Andersen on a Sunday morning and found her working in her laboratory. She listened to the medical history and saw his son the next day. The diagnosis was much more difficult and was made primarily on the clinical features plus analysis of the duodenal secretions. These were done and the diagnosis of cystic fibrosis of the pancreas was established. Since Dr Andersen was a pathologist she referred his son Lee for further clinical care to Dr Paul di Sant’Agnese. “He was a pediatric specialist and a delight – soft spoken, reassuring and supportive” (from Doershuk CF, 2001. below).
See further comments by Andersen (1939 below) describing how her interest developed and also suggesting the vitamin A deficiency resulting from the intestinal malabsorption was important in causing the general manifestations – later shown not to be the case.
1938 Blackfan KD, May CD. Inspissation of secretion and dilatation of ducts and acini, atrophy and fibrosis of the pancreas in infants. A clinical note. J Pediatr 1938; 13:627-634.
Examination of the pancreas in over 2,800 paediatric necropsies, performed over a period of 15 years in Boston, disclosed 35 examples of a well-defined pathologic lesion the existence of which was unsuspected during life. The lesion was characterized by inspissation of secretion, dilatation of the ducts and acini, atrophy and fibrosis. Only seven showed the stratified epithelium characteristic of vitamin A deficiency. The need for further information concerning the absorption of fat and especially nitrogen as well as pancreatic enzymes as an aid to diagnosis was suggested. The average age of death was 8 months.
Blackfan & Wolbach (1933 above) from a study of the lesion in the large amount of material available, in regard to pathogenesis, had observed “Our preliminary studies indicate that the pathogenesis of this striking pancreatic affection (figure 10) resides in the production of an abnormal secretion which inspissates and leads to distension and atrophy of the ducts and acini” (See also Blackfan & Wolbach, 1933 above; May CD 1943 below).
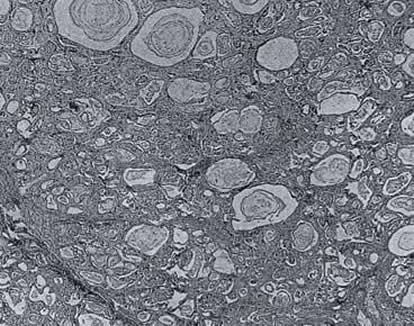
Fig. 13: Pancreas – Inspissated secretion, dilation of the ducts and acini, atrophy and fibrosis. From Blackfan & May, 1938 with permission

Charles May medicine.uiowa.edu
Dr. Charles D. May (1908 – 1992) was a paediatrician, an expert on childhood nutritional disorders and a leading figure in the United States paediatric world at the time. He qualified at Harvard Medical School in 1935. In World War II he served in the Army Medical Corps, returning to research at Harvard Medical School in 1946. May became an Associate Professor of Pediatrics at the University of Minnesota from 1947 to 1952, and subsequently held senior positions in Iowa, New York and Denver. He was succeeded as head of the Nutrition Clinic in Boston in 1947 by Harry Shwachman (of whom a great deal more later) who had worked with him before the war when May was the chief resident.
Shwachman many years later recalled that “May’s interest in CF was limited because he didn’t have a laboratory…and he wrote a textbook on CF because he wanted to write down his experience before he left the clinic. He went to England with an army unit and gave a lot of lectures in London at Great Ormond Street Clinic in London. And Dr Norman who was in charge of that clinic told me that he learned all he knew about CF from Dr May. And I had been May’s right hand man for a number of years so it was logical that I take over the clinic when he left” (Fanos JH. 2008 Am J Med Genet Part A 146A:284-293).[PubMed]
It is likely that May gave the first lecture on CF in the UK at the Royal Society of Medicine during his time in the US army (May, 1943 below). Later he held the Chair of Pediatrics at the University of Iowa where later he chaired the first scientific meeting of the National CF Research Foundation in 1955 which was attended by most of the few US paediatricians involved in CF care. Also, around 1953, he was one of the clinicians to observe that Pseudomonas aeruginosa was usually involved when persisting pulmonary infection was present. According to Warren Warwick, May also authored a seminal work on pancreatic enzymes.
More details of the life and career of Dr. Charles D May can be found in the following article – In Memoriam. Charles D May, MD. (1908-1992) by Renee K Bergner. Allergy Proc May-June 1993; 7(3)230-231.
1938 Davie TB. Massive replacement of the pancreas by adipose tissue. J Path Bact 1938; 46:473.
A severely retarded child who died at four years had massive fat replacement of the pancreas with sparing of the islands of Langerhans, considered to have been present since early infancy. Some patients with obstruction of the pancreatic ducts develop massive fat replacement and others show atrophy of the pancreas; it is not clear whether primarily due to obstruction of the pancreatic duct. The classical experiments of MacCallum ligating the pancreatic duct of dogs which eventually led to the isolation of insulin and the persistence of the islets were discussed (Banting & Best, 1922 above). Apparently liver failure is likely with massive fatty replacement if there is secondary infection. The author considered the present case supports the view that life could continue in the absence of exocrine pancreatic secretion.
1939 Andersen DH. Cystic fibrosis of the pancreas, vitamin A deficiency and bronchiectasis. J Pediatr 1939; 15:763-771.
This Mead Johnson Award lecture was presented before the American Academy in 1939. Dorothy Andersen’s 1938 paper (above) was attracting considerable attention and here she describes how she became involved in the study. Andersen notes that pancreatic deficiency in childhood had been considered infrequent but believed it was the recognition rather than the disease that was unusual!
Her interest had been aroused first by a child considered to have coeliac disease yet who died of bronchopneumonia following an operation; at autopsy the pancreas was found to be composed of small and large cysts and a few tubules embedded in a mass of fibrous tissue. She became convinced that the pancreatic lesion was the cause of the symptoms and so began a search for similar cases. In 1000 autopsies at the Babies Hospital, New York, the pancreas had been examined in 650 and in 20 this showed evidence of fibrosis; most had died of pneumonia aged less than 6 months, comparable to 8 month age of death of Blackfan & May’s series (1938 above). She obtained further cases from the literature and colleagues to a total of 49 which formed the basis of her 1938 seminal publication (Andersen, 1938 above).
Fourteen of the 49 children with CF that Andersen reported in 1938 showed histological evidence of vitamin A deficiency – in particular, epithelial changes characteristic of vitamin A deficiency. Also seven of 12 cases of vitamin A deficiency in the literature who had post mortems had pancreatic lesions (Blackfan & Wolbach, 1933 above).
For some years Andersen considered that many of the extra-gastrointestinal clinical manifestations of CF were due to vitamin A deficiency secondary to the severe malabsorption caused by the primary pancreatic abnormality. Andersen also emphasised the importance of demonstrating a very low titre or absence of trypsin from the duodenal juice in making the diagnosis.
It is stressed that at this time the cause of the condition was quite unknown and the hereditary aspects had not yet been recognised.
1939 Rauch S, Litvak AM, Steiner M. Congenital familial steatorrhoea with fibromatosis of pancreas and bronchiolectasis. J Pediatr 1939; 14:462-490.
These authors wrote – “We propose to describe what we consider a clinical entity. Because of its apparent familial incidence, its congenital nature, its association with respiratory symptoms, and the post-mortem findings we suggest the term congenital familial steatorrhoea with fibromatosis of the pancreas and bronchiolectasis”.
There is a very detailed account of previously reported cases with details of two new children who died aged seven months and two and a half years from bronchopneumonia. Post mortem on one showed the typical pancreatic changes of cystic fibrosis. The authors classified steatorrhoea into “idiopathic” which included true idiopathic celiac disease and the “pancreatogenous variety” as follows – “A review of the reported cases in the literature leaves the impression that steatorrhoea, excluding the acquired forms in adults of pancreatic inflammation or lithiasis, or those in which pathology of the lacteals is found, may be divided into idiopathic and pancreatogenous forms. The former includes celiac disease and sprue, tropical and non-tropical”.
This paper was obviously published at the same time as Andersen’s 1938 paper and the authors comment – “Since this paper was completed, the article of Dr Dorothy Andersen “Cystic fibrosis of the pancreas and its relation to celiac disease” (1938 above) and that of Dr John Thomas and Dr FM Schultz “Pancreatic steatorrhoea” appeared in the August 1938 issue of the American Journal of Diseases of Children” (1938 above).