The early years – “cystic fibrosis” by any other name


From the mid-17th century there were many reports of infants who may well have had cystic fibrosis by the nature of their clinical signs and course
1595 Professor Pieter Pauw (1564-1617)
The first description of the pancreas in a child who almost certainly died with cystic fibrosis (CF) is usually attributed to Professor Pieter Pauw (1564 -1617), the Professor of Botany and Anatomy at Leiden who wrote “On January 16th 1595, in a square leading to the Grand Canal in Treuendeel, in the presence of Drs Treloatius, Heurnius and Trutius, I conducted an autopsy on an 11-year old girl said to be bewitched. She had had strange symptoms for eight years. Inside the pericardium, the heart was floating in a poisonous liquid, sea green in colour. Death had been caused by the pancreas which was oddly swollen. It was very close to the rounded side of the liver, so that one could have thought, in touching it, that it was a scirrhous (a type of cancer with a hard and woody texture). When it was removed the interior was found to be brightly coloured, a kind of hard white viscous mass. The little girl was very thin, worn out by hectic fever (a fluctuating) but persistent fever”.

Fig. 2: Pieter Pauw performing an autopsy in the Anatomical Theatre in Leiden. From The Paradox of the Pancreas. Modlin IM, Kidd M (Eds). 2003:280. With the authors’ permission.
Pieter Pauw had been appointed in 1589 and from 1593, during the winters, he performed public postmortems. The “Theatrum anatomicum” accomodated up to 200 people – not only students but also surgeons and interested laymen.
1606 Alonso y de Los Ruyzes de Fontecha J. Diez previlegios para mugeres prenadas. L P Grande, Alcala de Henares, 1606, p 212. (figure 3)
The Professor of Medicine at Henares in Spain first described the salty taste of infants with cystic fibrosis. He wrote that it was known that the fingers tasted salty after rubbing the forehead of the bewitched child (Quoted by Quinton PM. Physiological basis of cystic fibrosis: a historical perspective. Physiol Rev 1999; 79:S3-S22) [PubMed]. Busch notes that reference to this finding could be found in the 19th century in eleven European states – in Poland, both German states, the Soviet Union, Czechoslovakia, Austria, Switzerland, Hungary, Romania, Yugoslavia and in Spain (Busch, 1987 below).

Fig. 3: The original Alonso y de Los Ruyzes de Fontecha document.
1729 Schmidt J G. Book of Folk Philosophy. Chemnitz, Joh Christoph and Joh David Stoseln.
It was stated that an excessively salty taste to the skin meant a child was bewitched.
(Quoted by Taussig LM (Ed.). Cystic Fibrosis. Thieme-Stratton Inc, New York. 1984).

Fig. 4: Sir Everard Home. Portrait from www.wikigallery.org/

Fig. 5: Karl Freiherr von Rokitansky. From Wikipedia
1813 Home Sir Everard. On the formation of fat in the intestines of living animals. Philos Trans Royal Soc (London) 1813; 103: 146-158.
Sir Everard Home (1756-1812) (figure 4) was a pupil of John Hunter whose sister he married. He worked with Hunter for many years. Among many appointments he was surgeon at St George’s Hospital, London after Hunter’s death in 1793. He described patients who had steatorrhoea considered to be due to pancreatic insufficiency.
1838 Rokitansky C von. Sections-Protokoll und Gutachen. Wien, 4. April 1838 (als Anhang)
Karl Freiherr von Rokitansky (1804-1878) (figure 5) was a distinguished Viennese pathologist and described as a founder of modern pathological anatomy. He was one of the towering figures who made the New Vienna School into a world medical centre in the second half of the nineteenth century. His contributions were fundamental to the establishment of pathology as a recognised science, and he himself performed more than 30,000 autopsies. He was one of the few who stood by the side of Semmelweis in the controversy over aseptic methods.
Rokitansky described the autopsy findings in a seven month gestation premature male fetus found in a box in a cemetery in Vienna in 1834 as follows – “Truncus foetus septimestris, in cujus abdomine intestinum ileum proxime valvulum coeci pariete convexo hiat foramina semen cannabis aequante tunicis intestine extus revolutis cincto in peritonaei cavum illincque meconium effudit”.[“In the last part of the small bowel there was a small ileal perforation the size of a hemp seed and around the perforation there was yellow jelly-like meconium partially fast sticking to the outside of the bowel”]. Death was considered due to peritonitis secondary to the ileal perforation. The report did not include microscopic description of the pancreas, although it was macroscopically normal.
Although the histology of the pancreas was not recorded, this report has been considered to be an early or even the first description of meconium ileus in cystic fibrosis. However, it was later shown that meconium ileus can occur in non-CF infants (Rickham et al,1965 below). Later it was the recognition of the changes in the pancreas, present in some of the many infants that came to autopsy, that led to the eventual recognition of “fibrocystic disease of the pancreas” as a specific entity by Dorothy Andersen in 1938 (below) even though the changes in the pancreas are very variable in severity.
1848 Pfyffer, J X. (zit bei 46): zitierend aus dem Wörterbuch der schweizerdeutschen Sprache 7 (1848).
From the The Dictionary of the Swiss-German Language -“If it tastes salty when someone is kissed on the brow, then this person is hexed” (bewitched).

Fig. 5A Ernst Ludwig Rochholz Wikipedia
1857 Rochholz EL. Alemannisches Kinderlied und Kinderspiel aus der Schweiz, In the Almanac of Children’s Songs and Games from Switzerland: Leipzig. JJ Weber, 1857. p280.
“The child will soon die whose brow tastes salty when kissed”. Yet another widely used quotation indicating that a salty taste on the skin indicated a poor prognosis with the implication that such a child may have had cystic fibrosis.
1888 Gee S. On the coeliac affection. St Bartholomew’s Hospital Report 1888; 24:17-20.
Samuel Gee (1839-1911), (figure 6) was physician to St Bartholomew’s Hospital and The Hospital for Sick Children, Great Ormond Street, London. In a lecture in 1887 Gee described a syndrome he termed the coeliac affection as follows –
“There is a kind of chronic indigestion which is met with in persons of all ages, yet is especially apt to affect children between one and five years old (figure 7). Signs of the disease are yielded by the fæces; being loose, not formed, but not watery; more bulky than the food taken would seem to account for; pale in colour, as if devoid of bile; yeasty, frothy, an appearance probably due to fermentation; stinking, stench often very great, the food having undergone putrefaction rather than concoction”.
The cause of the coeliac affection was unknown until the role of gluten was described by Willem Dicke in his MD thesis in 1950.

Fig. 6: Samuel Gee. From Wikipedia
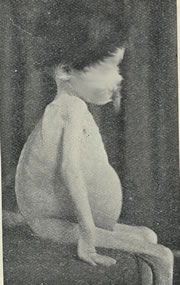
Fig. 7: Wasted boy with coeliac disease. Common Disorders and Diseases of Childhood. Still. 5th Edition, 1927.
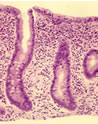
Fig. 8: Small bowel mucosa in coeliac disease showing subtotal villous atrophy.
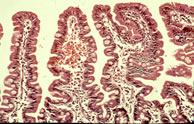
Fig. 9: Normal small bowel mucosa.
Eventually the two most important causes of this syndrome of the “coeliac affection” were identified as gluten-induced enteropathy, or coeliac disease as we know it today, and cystic fibrosis.
Subsequently two important advances in the Fifties permitted a clear separation of these two conditions. The first was the discovery of the elevated salt content of the sweat in CF reported in 1953 by Paul di Sant’Agnese from New York (di Sant’Agnese et al, 1953 below). The second was Willem Dicke’s discovery of the central role of “a factor in wheat” (gluten) in the aetiology of coeliac disease reported in his MD Thesis in 1950 (Dicke et al, 1953 below).
Subsequently the characteristic histological small bowel appearance of subtotal villous atrophy of gluten induced coeliac disease (figure 8) was first identified by Paulley in 1954 in laparotomy specimens from adults with idiopathic steatorrhoea (coeliac disease) (Paulley J W. Observations on the aetiology of idiopathic steatorrhoea. Jejunal and Lymph node biopsies. BMJ 1954; 173:1318-1321. [PubMed]) and
then in 1957 by Margot Shiner (figure 9a) in specimens obtained by per oral small bowel biopsy from an 8 year old child (Sakula J, Shiner M. Coeliac disease with atrophy of the small intestine mucosa. Lancet 1957; ii: 876-877. [PubMed]) and subsequently in children by Charlotte Anderson in 1960 (Anderson et al, Arch Dis Child 1960; 35:419-427. [PubMed]). In contrast, the intestinal villi are of normal height in people with CF – in some children with CF we found them to be even taller than normal (figure 9).

Fig. 9a Margot Shiner RCP London
Dr Margot Shiner (figure 9a) (1923–1998) was born in Berlin, Germany, to a cultivated merchant family that in the 1930s was obliged to flee Nazi persecution, first to Prague, then to London. Intent on a career in medicine, she completed her course at the medical school of Leeds University in 1947, then undertook specialized training in internal medicine and paediatric gastroenterology in London. She became a full-time member of the Medical Research Council’s gastroenterological unit at the Central Middlesex Hospital where she pursued seminal studies on the ultrastructure and cytopathology of the human small intestine. In 1956 she devised a jejunal biopsy tube based on the gastric mucosal biopsy instrument introduced a few years earlier by Ian Wood in Australia. By this means she clearly established the histopathologic identity of childhood and adult celiac disease.(information from RCP museum)
Although Sir Wilfred Sheldon and Eddie Tempany at Great Ormond Street considered the investigation to be “outside the scope of routine investigations in children” due to the increased difficulty and risk in small children (Sheldon W, Tempany E. Small intestine peroral biopsy in coeliac children. Gut 1966; 7:481-489), Professor John Walker-Smith believes that the development of the technique to perform oral small intestinal biopsy in childhood was the real beginning of paediatric gastroenterology (Walker-Smith JA. Historic notes in pediatric gastroenterology. J Pediatr Gastroenterol Nutr 1997; 25: 316). I would support this view (Littlewood JM. Coeliac disease in childhood. In: Howdle PD (Ed.). Clinical Gastroenterology. International Practice and Research. Bailliere Tindall, London. 1995; 9:295-328).
In the Sixties and Seventies paediatric subspecialities often developed on the back of a specialised investigation that a number of paediatricians learned from adult physicians. The first was obviously cardiac catheterisation by paediatric cardiologists then came various biopsy procedures including small intestinal biopsy during the Sixties and later fibreoptic endoscopy of the lungs and gastrointestinal tract.
1900 Arraga A, Vinas M. Sclerosis of the pancreas accompanied by chronic gastroenteritis. Arch Med Enfant Paris 1900; i: 1497-1500.
Ten children with gastroenteritis were found at autopsy to have histological changes in the pancreas with inflammation causing sclerosis with possible blockage of the main pancreatic duct of Wirsung.
1904 Bramwell B. Pancreatic infantilism: remarkable improvement (growth of body and sexual development) as a result of the administration of pancreatic extract. Trans Medico-chi Soc Edin 1904; 23; 162. Clinical Studies 1904; 2:348-352. Edinburgh. R and R Clark.
Dr Byrom Bramwell (1847-1931) (figure 9b) notes the previously described association of chronic diarrhoea and arrested body development. He describes a boy aged 18 years who was the size of one aged 11 years, whose size and sexual development responded to one drachm of “Armour’s liquor pancreaticus” and one drachm of glycerine extract of “steapsin” three times daily.
Bramwell writes :- “After carefully studying the case I came to the conclusion that the diarrhoea was due to defective metabolism in the upper part of the gastrointestinal tract: and I suggested that it was probably due to disease or defective action of the pancreas”.
Pancreatic secretion was shown to be defective by demonstrating undigested fat in the stool, by a low phosphoric acid content of the urine after administration of milk and by Sahli’s test of digesting a glutoid capsule surrounding iodoform and demonstrating the released iodine in the saliva.
Bramwell published six other papers on “pancreatic infantilism” between 1902 and 1915 at various Edinburgh meetings and certainly some of these appeared to be recording progress of the same patient.
In this present report Bramwell writes :–
“I claim that there is a distinct variety or form of infantilism which is due to disease of the pancreas and that this, the pancreatic form of infantilism, as I have ventured to term it, can be cured by administration of pancreatic extract. I consider that it is a distinct clinical entity – a disease which has not hitherto been recognised or described”.
So this is probably an early description of cystic fibrosis as a distinct clinical entity! Leonard Parsons considered, almost certainly incorrectly, that some of these cases were examples of coeliac disease (Parsons, 1935 below; comment by Rentoul, 1904 below). Admittedly their length of survival is against their having CF although it is possible they had an atypical form caused by milder genetic mutations.

Fig. 9b: Byrom Bramwell. RCP Edinburgh Library with permission

Fig. 9c: John Thomson. RCP Edinburgh Library – with permission
1904 Thomson J. Exhibition of patients: two cases of infantilism. Trans Med Chirg Edinburgh 1904; 23:165-166.
Dr John Thomson of Edinburgh (1856-1926) (figure 9c) “exhibited two cases of infantilism accompanied by severe dyspepsia and diarrhoea of many years’ duration, and probably due to some morbid condition of the pancreas”.
One a man aged 24 years was the height of a boy aged nine years (51.5 inches), he had weak muscles and a tumid abdomen. He had been well until severe influenza at the age of 11 years was followed by intractable diarrhoea. There was very slow growth and a wide variety of treatments were of little value.
A similar young man aged 17 years (height on 49.5 inches – the average height of an 8 year old boy) was pre-pubertal and had chronic diarrhoea. The diarrhoea in both these patients was considered to be pancreatic in origin and the clinical features similar to those patients of Dr Byrom Bramwell (1904 above) but no evidence to support this assumption was presented.
Dr John Thomson (1856-1926) was described by the late Professor William Craig as “Pioneer and Father of Scottish Paediatrics” in his monograph (WS Craig. Edinburgh & London:E & S Livingstone 1968). In a review of Craig’s book TE Cone notes that “the book is obviously a labour of love because the author, as did everyone else who knew Thomson, was deeply inspired by his gentleness, his kindness, and his ungrudging, unsparing, and unyielding efforts to improve the care and cure of children”.
1904 Rentoul JL. Pancreatic infantilism. Brit Med J 1904; 2:1694-1695.
A patient apparently diagnosed by Dr Rentoul of Lisburn, Co Antrim, thanks to Byron Bramwell’s previous descriptions of pancreatic infantilism (Bramwell, 1904 above). Rentoul described a girl aged 18 years with severe growth retardation so she looked more like a nine year old and also had a swollen abdomen. There had been lifelong chronic diarrhoea that responded impressively to pancreatic extract with improved growth and signs of puberty but whose bowel symptoms relapsed when this was withdrawn. In four months on the pancreatic extract she gained 9.5 lbs in weight and gained 2 inches in height; also on pancreatin she was “bright happy and willing for any work”!
The cases reported above by Bramwell 1904, Thomson, 1904 and this of Rentoul could have had Shwachman-Diamond syndrome (Shwachman et al, 1964 below) or congenital lipase deficiency (Sheldon W. Arch Dis Child 1964; 39:268-271. [PubMed]; Ligumsky M et al. Gut 1990; 31:1416-1418. [PubMed]) as chest involvement did not seem to be a significant feature and also the age of presentation was far beyond the likely survival age of a child with CF at that time.
1905 Landsteiner K. Intestinal obstruction from thickened meconium. Zentralbl Allg Pathol 1905; 16:903-907.
This was the first description of an infant with meconium ileus to be accompanied by a description of the associated pancreatic histological changes. The pancreas showed an increase in inter and intra lobular connective tissue and round cell infiltration and markedly dilated ducts. Landsteiner suggested that lack of pancreatic secretion had caused the thickening of the meconium. He mentions another infant where the pancreas was normal but there was no secretion of bile. The report is considered to be the first to suspect that microscopic changes in the pancreas could interfere with secretions and digestion of meconium and thus result in bowel obstruction. Oppenheimer & Esterly, the distinguished N. American pathologists, regard this as the first report of meconium ileus (1975, below)

Fig. 10: Karl Landsteiner. From www.nndb.com
Karl Landsteiner (1868-1943) (figure 10) was a Viennese pathologist who worked first in Vienna then from 1922 in the United States. He described the blood iso-agglutinins in 1900 and the blood groups in 1901 for which he received the Nobel Prize in Physiology of Medicine in 1930. He also, with Alexander Wiener, discovered the Rhesus factor in 1937.
Prolapse of the rectum (figure 10a) is a common disorder among children, writes Mr. Lockhart-Mummery a distinguished London surgeon and pioneer in cancer surgery, “especially that class which attends the surgical practice at a children’s hospital”. He had seen examples of the usually quoted causes of rectal prolapse (phimosis, stone in the bladder, colonic polyps, etc) to be rare but chronic diarrhoea, wasting and general weakness to be common – particularly the removal of fat from around the rectum in such cases.
Unfortunately there is no mention of the long term outlook of these children that may have suggested some may have had CF; however, the fact that the average age was 2.56 years perhaps indicated that prior to 1907 most children that had a prolapse due to CF would have already died. Also in 1907 death in early childhood from gastroenteritis was relatively common as was malnutrition and wasting.
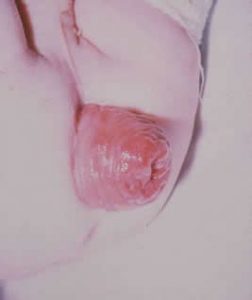
Fig Severe rectal prolapse

Fig. 11: Christian A Herter. From Wikipedia

Fig. 12: Otto Heubner. From Wikipedia

Fig. 13: Sir Archibald Garrod. From
Subsequently rectal prolapse was described as a definite feature of CF (Kulczycki & Shwachman, 1958 below). The lack of fat around the rectum, as suggested by Lockhart-Mummery, is obviously relevant in CF for when this is restored with appropriate pancreatic enzyme treatment the tendency to rectal prolapse diminishes – usually completely.
In over 600 young people receiving treatment for CF I recall only one teenage girl who still complained of occaisional rectal prolapse. However, one wonders if persistence of this embarrassing problem is under-reported as was later found to be the case with urinary incontinence in people with cystic fibrosis (Cornacchia et al, 2001 below).
1908 Herter CA. On infantilism from chronic intestinal infection. MacMillan, New York. 1908:18.
Christian Archibald Herter (1865-1910) (figure 11) from New York was a pathologist and an expert on metabolism who had qualified in medicine at the age of 18 years!
He described “intestinal infantilism due to chronic intestinal infection causing an unsuitable intestinal bacterial flora resulting in excessive putrefaction and chronic intoxication of the neuromuscular system” – subsequently known as Herter’s Disease. This appeared to be the same clinical condition as Samuel Gee had described in 1888. Some children did well for the first year or so then developed chronic gastrointestinal symptoms and signs and growth problems, steatorrhoea and altered faecal bacterial flora. There were no post-mortems so the state of the pancreas was unknown.
The fact that Herter’s cases were aged eight, nine, four, three and nine and a half years makes it very unlikely they had CF, as few children with CF survived to these ages at that time, and so they were more likely to have had coeliac disease.
1909 Heubner O. Serious digestive insufficiency of children beyond infancy. Jahrb Kinderh 1909; 70:667-700.
Johan O L Heubner (1843-1926) (figure 12) was Professor of Paediatrics in Berlin and Leipzig. He studied infant nutrition and warned against the prolonged sterilisation of milk. He described the infantile form of idiopathic steatorrhoea – subsequently this became known as Heubner-Herter disease (Herter, 1908 above). Heubner also isolated meningococci from the spinal fluid and described syphilitic endarteritis of the cerebral vessels.
1911 Freeman RG. The intestinal infantilism of Herter. Am J Dis Child 1911; 2:332-339.
Freeman (figure 16a) discusses Herter’s 1908 cases (Herter, 1908 above) and notes recent attention has been given to classifying the different types of “dwarfs” described as intestinal (Herter) hepatic (Coutley), and pancreatic (Bramwell).
The discussion implies that Herter described a specific condition which is unlikely to be the case.
In this paper Freeman describes four patients, aged between two years five months and four years six months with chronic gastrointestinal symptoms two of whom improved with pancreatic extract – “Have good appetite, take food eagerly but have the large poorly digested movements with a great waste of fat”. There were no postmortems as all four children gradually improved and appeared to survive which makes CF an unlikely diagnosis. (also Freeman 1922 below)
1912 Garrod AE, Hurtley WH. Congenital familial steatorrhoea. Q J Med 1912; 6:242-258.
Sir Archibald Garrod (1857-1936) (figure 13) was a London physician and an expert in inborn errors of metabolism. He succeeded Sir William Osler as Regius Professor of Medicine at Oxford and wrote his famous “The Inborn Errors of Metabolism” in 1931.
Garrod notes that the term “steatorrhoea” was first employed by Kunzmann in 1824 to designate the passage of liquid fat in the stools but later, rather confusingly, the term was applied by Sir James Erasmus Wilson (1809-1884), a distinguished London dermatologist, to the disease of the skin commonly known as seborrhoea! Garrod reviews the details of previously published families some of whose children had steatorrhoea and died of pneumonia suggesting a recessive inheritance.
The present report describes a well-grown child aged eight years who “passed grease” from the bowel – a sign very suggestive of pancreatic insufficiency. He was the second of five children of whom only two survived – one had died of pneumonia at seven months, one at six weeks with convulsions and the third at eleven months with measles and bronchopneumonia. The parents were first cousins. The boy’s stools contained “large quantities of liquid fat which solidified on cooling to form a plate around the faecal mass. Liquid fat escaped involuntarily from the anus at times”. A variety of investigations suggested there was no overall defect of pancreatic secretion “as a whole” suggesting “an inborn error hitherto undescribed” – suggesting “that in congenital steatorrhoea some agent which facilitates absorption is, in like manner (to alkaptonuria), wanting”. He suggested “An error of fat absorption accompanied by, but not dependent on, a limitation of the power to split fats”.
Despite the family history, the overall progress, normal growth, lack of respiratory problems, presence of tryptic digestion are against this child having CF, but could indicate he had “congenital lipase deficiency”, a condition later described by Sir Wilfred Sheldon, paediatrician at Great Ormond Street, London in two families, each with two affected children (Arch Dis Child 1964; 39:268-271). Sheldon’s patients had severe steatorrhoea and a tendency for oil to ooze from the anus, but satisfactory growth, absent or low pancreatic lipase but normal amylase and proteolytic enzymes in the pancreatic juice. (Also Garrod AE. Congenital family steatorrhoea. BMJ 1920; i: 249-264).
1914 Poynton FJ, Armstrong RR, Nabarro DN. Contribution to study of a group of cases of recurrent diarrhoea in childhood. Brit J Child 1914; 11:145-155 and 193 –201.
While studying the chronic diarrhoeas of childhood the authors noted marked increase in interlobular fibrous tissue of the pancreas, particularly surrounding the ducts, in one of their fatal cases of “coeliac disease” – it is likely that this child had cystic fibrosis. The authors suggested two factors in the aetiology of the disease – a possible infection and the appearance of the coeliac affection symptoms as sequelae to the pancreatic damage.

Fig. 14: Dr Frederick J Poynton. (1869-1943) From HHARP website – permission requested

Fig. 15: Sir George Frederick Still. Reproduced with permission from Corporate Communications, King’s College Hospital NHS Foundation Trust
Dr Frederick J Poynton (1869-1943) (figure 14) was physician at Great Ormond Street from 1893 to 1934 and President of the British Paediatric Association in 1931.The two volumes of unpublished memoirs compiled by him after his retirement and held by the Hospital Archives contain a host of interesting information about day to day life at the Hospital, developments in clinical practice, and personal observations on the pioneering generation of physicians and surgeons working at Great Ormond Street in the late 19th and early 20th centuries. They included Thomas Barlow, Frederic Still, F E Batten, Archibald Garrod, Edmund Owen and William Arbuthnot Lane. The memoirs also throw critical light on children’s nursing in that era, the growing pressures on the Voluntary Hospital system that ultimately resulted in the creation of the NHS, and the emergence of specialist Paediatrics (and sub-disciplines within it) by the 1930’s. (from a presentation by Nick Baldwin, GOS Archivist, at the British Society for the History of Paediatrics and Child Health, 2010)
1918 Still GF. Lumleian lectures on Coeliac Disease. Lancet 1918; 2:162 and Lancet 1918; 2:193.
Sir George Frederick Still (1868-1941) (figure 15) noted the majority of his cases of coeliac disease occurred in the later part of infancy, none ever beginning while the child was breast fed. He had seen 17 such children amongst 14,800 hospital patients but 24 in his private practice. One of his cases had loose stools for four months at seven years and died aged seven and a half years and had excess fibrous tissue especially about the ducts suggestive of pancreatitis. He explained the presence of pancreatitis and round cell infiltration of the intestinal mucosa and submucosal in one case as an inflammatory process secondary to some functional failure of digestive secretion with resulting abnormal decomposition of food residue favouring growth of harmful bacteria.
This child is not mentioned in Still’s classic paediatric textbook “Common Disorders and Diseases of Childhood” 1927 edition. The only brief mention of the pancreas is in the chapter on coeliac disease – “On the assumption that some defect of the pancreatic secretion underlies coeliac disease, a view put forward by Dr Cheadle and others, various pancreatic extracts have been tried. I can only say that in my experience such drugs have had very little if any effect, certainly none comparable to the effect of the castor oil mixture”. So there was no suggestion that some of these children may have suffered from pancreatic abnormalities. Also the ages and outlook of the children would be against their having cystic fibrosis.
1919 Passini F. Pankreaserkrankung als Ursache des Nichtgedeihens von Kindern. (Pancreatic disease as a cause of failure to thrive in children) Deutsche Med Wschr 1919; 45: 851-853
Passini described pancreatic disease in an infant aged two months with nutritional failure and large stools. The pancreatic histology was typical of that subsequently described in cystic fibrosis; he also described a sibling, aged nine months, who had a small pancreas with cystic changes in the acini and a reduction in the islets of Langerhans. Another infant aged 18 months showed widening of the pancreatic ducts and a necrotic parenchyma. All died of bronchopneumonia and presumably all had cystic fibrosis.
Martin Bodian (see 1952 below), pathologist at the Hospital for Sick Children, Great Ormond Street, London and other authors (for example Wolman 1942), considered that Passini was the first to suggest and prove the presence of a definite abnormality of the pancreas in some cases of steatorrhoea which he differentiated from Herter’s disease – chronic intestinal infantilism possibly due to some disturbed bacterial situation in the gut (Herter, 1908 above).

Fig. 16: Sir Frederick Banting. From Wikipedia
Miller described a child with the clinical features of cystic fibrosis who died at home following a dental extraction. There was 52 percent of fat in the stools but there was no autopsy. Later Miller reviewed the post mortem findings of some previous cases and concluded that the excessive fat loss in the stools was due to enteritis and must be due to a digestive fault, probably a defective action of bile salts (Miller R. Lancet 1921; i: 743; Miller R. Arch Pediat 1923; 40:88).
1922 Banting FG, Best CH. Internal secretion of the pancreas. J Lab Clin Med 1922; VII: 251-266.
The work reported in this classic paper eventually led to a Nobel Prize in 1923 for Banting and Macleod, in whose Toronto laboratory the work was done. Frederick Banting (1891-1941) (figure 16) was a Canadian orthopaedic surgeon who became an assistant in physiology in Ontario where he worked in Richard MacLeod’s laboratory with Charles Best, a medical student. It was here, after many ups and downs and arguments and with the help of Bertram Collip, a highly trained biochemist (to extract the insulin) that they eventually produced and tried the insulin on a diabetic patient in 1922.
The paper begins –“The hypothesis underlying this series of experiments was first formulated by one of us in November 1920 (Banting was then assistant in Physiology at Western University, London, Ontario) while reading an article dealing with the relation of the isles of Langerhans to diabetes (Barron M: The relation of the islets of Langerhans to diabetes with special reference to cases of pancreatic lithiasis. Surg Gynec Obstetr 1920; xxxi :437-448). From the passage in the article which describes the degenerative changes in the acini of the pancreas following ligation of the ducts, the idea presented itself that since the acinous but not the island tissue degenerates after this operation, advantage might be taken of this fact to prepare an active extract of the islet tissue. The subsidiary hypothesis was that trypsinogen or its derivatives was antagonistic to the internal secretion of the gland. The failures of other investigators in this much worked field were thus accounted for”.
The authors concluded from their subsequent experiments that –“intravenous injections of extract from dog’s pancreas, removed from 7 to 10 weeks after ligation of the ducts, invariably exercises a reducing influence upon the percentage sugar of the blood and the amount of sugar excreted in the urine”

Moses Barron Minnesota Historical Society
This Banting and Best paper is a medical classic and a “good read” and one of the most significant medical papers of the 20th century as also is Moses Barron’s paper that gave Banting the idea (mentioned in the quotation above).
Dr. Moses Barron (1884-1974) received his medical degree from the University of Minnesota Twin Cities in 1911, and later became Chief of Staff at Mt. Sinai Hospital, the only local hospital that consistently allowed Jewish doctors to practice medicine. In 1920, Dr. Barron published an article, “The Relation of the Islets of Langerhans to Diabetes,” which noted the importance of pancreatic cells in relation to diabetes. His research inspired others (Banting and Best) to look into the disease, leading to the Nobel Prize winning discovery of insulin as a treatment for diabetes in 1923.
Banting and Best’s paper is included here not only for its historical interest but also for its relevance to CF as the majority of those affected will eventually develop CF related diabetes in adult life. Although the Islets of Langerhans in CF are functioning adequately through childhood in most patients, despite their severe exocrine pancreatic insufficiency, they are eventually destroyed resulting in the majority of adults with CF developing diabetes mellitus. Efforts to prevent the slow destruction of the pancreas and prevent or delay the onset of CF related diabetes will surely become an area of research as more people with CF survive to develop this complication which has an adverse effect both on their prognosis and quality of life.
It is encouraging that in a small study of CF patients with a G551D mutation treated with ivacaftor there was a significant increase in insulin production in response to a glucose load (Bellin et al, 2013 below). Subsequent publications over the next decade confirmed the favourable effects of the new modulator drugs on CF diabetes.
Rowland Freeman of New York (1859-1945) (figure 16a) , in a discussion on celiac disease at the American Pediatric Society in 1922, recalled he had reported five children (Freeman, 1911 above) one of whom had very defective bone formation and various fractures. He had noted that “she seemed to derive more benefit from pancreatic extract than anything else”. Others at the meeting emphasised the importance of considering coeliac disease as a symptom complex rather than a specific condition although others disagreed and regarded it a distinct clinical entity.
So there was still considerable confusion regarding the specific causes of the malabsorption syndrome in childhood.

Fig. 16a: Rowland G Freeman. From Ancestry.co.uk
1923 Schick B, Wagner R. Über eine Verdauungsstörung jenseits des Säuglingsalters. (Atrophia pluriglandularis digestiva). Ztschr f Kinder 1923; 35:263-274.
A description of a disturbance of digestion after infancy due to “multiple glandular atrophies”. A child aged 16 months is described with atrophy of the pancreas in addition to “atrophy of other digestive glands and organs of internal secretion”. Also four other patients with similar features were described. Oedema was a particular feature in some. The authors suggested that in some infants with coeliac disease it was “an anatomically proved fact that glands of internal secretion which are in close relationship to digestion are primarily atrophic” and coined the term “atrophica pluriglandularis digestiva” (Hess & Saphir, 1935. below).
1923 Wilson JR, DuBois RO. Report of a fatal case of keratomalacia in an infant with postmortem examination. Am J Dis Child 1923; 26:431-446.
A female infant aged five months died 18 days after admission. She had been fed almost entirely on diluted condensed milk and was severely wasted. There was severe keratomalacia and eventually perforation of the left cornea. At autopsy microscopic examination showed extensive changes in many organs including the lungs, salivary glands and pancreas. There were inflammatory lesions in the lachrymal and salivary glands.
The pancreas (figure 17) showed keratinisation in certain ducts, many epithelial lined cyst like cavities, a marked inflammatory process and extensive fibrosis. The lungs also showed keratinisation of the epithelium, a marked peribronchitis, bronchiectatic cavities and abscesses. The authors observed – “To the mechanical effect of desquamated keratinized epithelium we are inclined to attribute the pancreatic cysts and bronchiectasis”.
This is an interesting paper as Dorothy Andersen, who described CF in 1938 (below), for many years considered the pancreatic lesions and intestinal malabsorption to be primary and the other systemic effects to be the result of the vitamin A deficiency which, for example, caused secondary squamous conversion of the lining respiratory epithelium. It seems almost certain that the present infant had CF with severe secondary vitamin A deficiency.
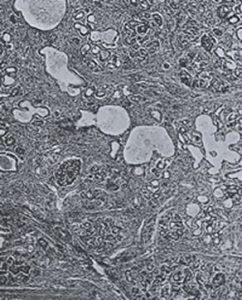
Fig 17. Histology of the pancreas from Wilson & Bubois, 1923,
1924 Clarke C, Hadfield G. Congenital pancreatic disease with infantilism. Q J Med 1924; 17:358 – 364.
A girl aged four years was admitted to the General Hospital in Bristol, UK in 1921 with fatty diarrhoea from birth. She was undersized, under weight, passed large bulky unformed and obviously fatty stools. She had never been jaundiced although the liver was greatly enlarged. She died from bronchopneumonia seven weeks later. Two of her eight siblings had died in infancy of bronchopneumonia. At autopsy there were miliary lung abscesses, atrophy and fibrosis of the pancreas – a shrunken looking dead white fatty mass occupied the position of the pancreas and the liver showed high grade fatty infiltration. “Histology of the pancreas yielded a striking but indirect confirmation of modern experimental and clinical knowledge of the function of the pancreatic islet tissue”.
Histology of the pancreas was typical of that subsequently described in cystic fibrosis (similar to that reported by Davie 1938, below).
1925 Mautner H. The Herter-Heubner digestive insufficiency. Klin Wchnschr 1925; 4: 164
A child with recurrent diarrhoea from the age of one year died from pneumonia at three years. There was high grade degeneration of the exocrine pancreas with the alveoli distended and filled with eosin rich secretion; the islets of Langerhans were intact. There was an enormous fatty liver and acute lobular pneumonia.
History and pancreatic findings were very suggestive of cystic fibrosis.
1925 Burghard E. Pankeaserkrankungen im Säuglingsalter. (Disease of the pancreas in infancy). Klin Wschr 1925; 4:2305-2306.
An infant who had failure to thrive and up to six foul fatty stools daily died at ten months with bronchitis and pneumonia. Pancreatic histology showed cystic degeneration and increase in interlobular connective tissue that was typical of cystic fibrosis.
1926 Thoenes F. Familial pancreatic insufficiency. Monatsschr Kinderheilkd 1926; 34:398-400.
A child aged one year and six months passed oily stools – “diese butterstuhle” regarded by Adolf Schmidt as pathognomonic of pancreatic insufficiency in adults. The parents were cousins and one sibling was retarded and another had died of pneumonia.
The character of the stools – particularly “oil”, or “butterstuhle” – is emphasised by some later authors as being very suggestive of pancreatic disease. Leakage of oil from the anus is also virtually diagnostic of pancreatic insufficiency.
1926 Gross F. Pancreatic atrophy in infancy and childhood. Jahrb F Kinder 1926; 112:251-257.
An infant aged three months with cachexia and bronchitis diagnosed as Heubner-Herter disease (infantile coeliac disease 1908, 1909 above) died of pneumonia. At autopsy there was pancreatic atrophy with replacement by fat and an increase in the number of islets. The cases where pancreatic disease seems to have been a major factor are reviewed; in particular the author draws attention to a paper by Nakamura. Untersuchungen uber das Pankreas bei foten, neugeborenen, kinder und im pubertatsalter. Virch Arch 1924; 253-286.
1927 Lehndorff H, Mautner H. Die Coeliakie. Herter’s intestinal infantilism, Heubner’s severe intestinal insufficiency beyond infancy. In Kraus F, et al. eds. Ergebnisse der inneren Medizin: Kinderheilkunde 1927; 31:456-593.
This long very detailed 137 page chapter starts with six pages of references and an historical review of Herter’s intestinal infantilism and Heubner’s so-called severe digestive insufficiency following infancy. The authors consider that Gee’s name should be attached to the condition in view of his 1888 description (Gee 1888, above) and that the term coeliac disease (Coeliakie in German) to be the best term.
The bulk of the chapter consists of a compendium of cases with their clinical details. The pancreatic findings are described – a case is described with lymphocytic infiltration of the stroma, distended glandular alveoli with reduction in the size of the glandular epithelial cells but filled with Sudan positive granules – the islets of Langerhans seem enlarged but are normal. The changes are considered to be secondary to the poor nutritional state. (The histology does not appear suggestive of CF and the cases described would have been unlikely to survive at that time had they had cystic fibrosis).
The authors concluded “The outcome of our work confirms that numerous investigations of many authors have failed to discover the origin of coeliac disease. The development and the cure still present unanswered problems. The symptomatology however appears to us so well worked out that one is justified to think of coeliac disease as a clinically sharply defined independent syndrome”. A view which others were to question over the next decade.
1927 Socknick A, Thoenes F. Familial pancreatic insufficiency, a contribution to pancreatic pathology in early childhood. Jahr Kinderheilk 1927; 115:315.
The authors tried to rely on a clinical differentiation of between coeliac disease and pancreatic insufficiency. In the former they found marked fat loss in the stool, slight protein intolerance, neuropathy, short stature, hydrolability, marked carbohydrate intolerance and increased peristalsis; in the latter, pancreatic insufficiency, there were “butter stools”, marked protein intolerance, no neuropathy or dwarfism, hydrostability, good carbohydrate tolerance and normal gut motility.
This appears to be a reasonable separation of coeliac disease and pancreatic steatorrhoea on clinical grounds although growth problems are a major feature of cystic fibrosis.
1927 De Lange C. Cirrhosis of pancreas and liver in infant. Am J Dis Child 1927; 34:372-383
Cornelia de Lange, famous for her description of the syndrome of multiple congenital anomalies which bears her name, described the third child of healthy parents who was jaundiced from the 7th day of life and admitted to hospital aged six weeks weighing only 2200 gm. The child appeared atrophic with peeling skin and hepatosplenomegaly and died two days later. At autopsy the pancreas showed a great increase in connective tissue and round cells but “excretory ducts showed nothing of importance”. Some islets were of abnormal size and sclerotic. The liver showed bile thrombi and evidence of obstruction. The heart and lungs were normal.
It is difficult to accept that this infant definitely had CF, but there was neonatal obstructive jaundice and histological abnormalities of the pancreas and so CF would be a definite possibility.

Fig. 18: Sir Alexander Flemming. From scotlandvacations.com with permission

Fig. 19: Sir Howard Florey From Wikipedia

Fig. 20: Ernst Boris Chain From Wikipedia

Fig. 21: Ernest Duchesne. From Wikipedia
1929 Kornblith BA, Otani S. Meconium ileus with congenital stenosis of the main pancreatic duct. Am J Path 1929; 5:249 – 261.
Stenosis of the proximal end of Wirsung’s duct (the main pancreatic duct) was found in an infant with meconium ileus who died aged five days. There was marked increase in the stroma of the pancreas; the acini were atrophic and many areas replaced by connective tissue. The main duct was extremely widened which suggested a congenital anomaly of duct development as a cause of the pancreatic fibrosis which was present.
Almost certainly CF in view of the pancreatic changes and the meconium ileus (Dodd, 1936 below; Landsteiner, 1905 above are similar).
1929 Flemming A. “On the antibacterial action of cultures of a penicillium, with special reference to their use in the isolation of B. influenza.” Br J Exp Pathol 1929; 10: 226–36.
Alexander Flemming (1881-1955) (figure 18) was a Scottish bacteriologist, working at St Mary’s Hospital, London, whose discovery of penicillin (1928) prepared the initial step towards the highly effective practice of antibiotic therapy for infectious diseases.
In 1945 Fleming shared the Nobel Prize for Physiology or Medicine with Ernst Boris Chain (1906-1979) (figure 20) and Howard Walter Florey (1898-1968) (figure 19) who both (from 1939) were responsible for carrying forward Fleming’s initial observation by further isolation, purification, testing, and quantity production of penicillin
In 1940 a report was issued describing how penicillin had been found to be a chemotherapeutic agent capable of killing sensitive germs in the living body. Thereafter great efforts were made, with government assistance, to enable sufficient quantities of the drug to be made for use in World War II to treat servicemen with war wounds. Penicillin became available for a few patients with CF in the USA in 1943 and the results were reported by Paul di Sant’Agnese (1944 below) – prior to this virtually all children with CF died in infancy or early childhood from Staphylococcal pneumonia and malnutrition.
It is noteworthy that Flemming failed to develop his 1929 discovery – this was done over 10 years later by Florey and Chain. Nor was Flemming the first to observe the antibacterial effects of moulds. Ernest Duchesne (1874 –1912) (figure 21) was a French physician who noted that certain moulds kill bacteria. He made this discovery thirty-two years before Alexander Fleming observed the antibiotic properties of penicillin, a substance derived from those moulds, but his research went unnoticed. Duchesne entered l’Ecole du Service de Santé Militae de Lyon (the Military Health Service School of Lyon) in 1894. Duchesne’s thesis, “Contribution à l’étude de la concurrence vitale chez les micro-organismes: antagonisme entre les moisissures et les microbes” (Contribution to the study of vital competition in micro-organisms: antagonism between moulds and microbes), that he submitted in 1897 to get his doctorate degree, was the first study to consider the therapeutic capabilities of moulds resulting from their anti-microbial activity.
Duchesne had made his breakthrough by observing how the Arab stable boys at the army hospital kept their saddles in a dark and damp room to encourage mould to grow on them. When he asked why, they told him that the mould helped to heal the saddle sores on the horses. Intrigued, Duchesne prepared a solution of the mould and injected it into a series of diseased guinea pigs. All recovered.
In a series of meticulous experiments, Duchesne studied the interaction between Escherichia coli and Penicillium glaucum, showing that the latter was able to completely eliminate the former in a culture containing only these two organisms. He also showed that an animal inoculated with a normally lethal dose of typhoid bacilli would be free of the disease if the animal was also inoculated with Penicillium glaucum. Unfortunately, as he was only 23 years old and unknown, the Institut Pasteur did not even acknowledge receipt of his dissertation! He urged more research but unfortunately his army service, after getting his degree, prevented him from doing any further work.
Considerable attention has been allotted to penicillin in this History as before the availability of penicillin few infants with CF survived beyond infancy. It was the introduction of penicillin and later other antibiotics which was the main development that permitted survival beyond infancy.