
History – 2018 (Section 1)


2018 A
THE FIRST THREE ABSTRACTS ARE REVIEWS OF VARIOUS ASPECTS OF PROGRESS IN 2018
Adrienne P Savant, Susanna A McColley.Cystic fibrosis year in review 2018, part 1.Pediatr Pulmonol 2019 Aug;54(8):1117-1128. doi: 10.1002/ppul.24361.Epub 2019 May 20. [Pubmed]
Cystic fibrosis research and case reports were robust in the year 2018. This report summarizes research and cases related to Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) modulator therapies, inflammation and infection, epidemiology and the physiologic, and imaging assessment of disease.
Adrienne P Savant, Susanna A McColley. Cystic fibrosis year in review 2018, part 2. Pediatr Pulmonol 2019 Aug;54(8):1129-1140.doi: 10.1002/ppul.24365.Epub 2019 May 24. Full text available [Pubmed]
Cystic fibrosis (CF) research and case reports were robust in the year 2018. This report summarizes publications related the multisystem effects of CF, pulmonary exacerbations, new and expanded therapies other than cystic fibrosis transmembrane conductance regulator modulator studies, and patient-reported priorities and outcomes.
The authors are in the Division of Pulmonary Medicine, Ann & Robert H. Lurie Children’s Hospital of Chicago, Chicago, Illinois.Department of Pediatrics, Northwestern University Feinberg School of Medicine, Chicago, Illinois.and susanna McColley also at the Stanley Manne Children’s Research Institute, Chicago, Illinois.
I M Balfour-Lynn. Clinical papers of the year 2018 – Cystic fibrosis. Paediatr Respir Rev 2020 Feb;33:58-61.doi: 10.1016/j.prrv.2019.03.008.Epub 2019 Apr 5. [Pubmed]
This paper reviews the most important clinical papers in cystic fibrosis he published in 2018, having searched all the literature on Pubmed. Focus is on CFTR modulator therapy, randomised controlled trials, and infection/microbiology issues.
Dr Ian Balfour-Lynn is in the Department of Paediatric Respiratory Medicine, Royal Brompton Hospital, UK. Electronic address: i.balfourlynn@ic.ac.uk.
2018 First author initial A to S
Abou Alaiwa MH, Launspach JL, Grogan B, Carter S, Zabner J, Stoltz DA, Singh PK, McKone EF, Welsh MJ Ivacaftor-induced sweat chloride reductions correlate with increases in airway surface liquid pH in cystic fibrosis.JCI Insight. 2018 Aug 9;3(15). pii: 121468. doi: 10.1172/jci.insight.121468. [Epub ahead of print] [Pubmed]

Fig. 1 Abou Alaiwa~Mahmoud
medicine.uiowa.edu
Compared with that in the newborn period, ASL pH increased by 6 months of age. In people with CF bearing G551D or R117H mutations, ivacaftor did not change the average ASL pH; however reductions in sweat Cl- concentration correlated with elevations of ASL pH. Reductions in sweat Cl- concentration also correlated with improvements in pulmonary function.
These results suggest that CFTR-independent mechanisms increase ASL pH in people with CF. the authors speculate that CF airway disease, which begins soon after birth, is responsible for the adaptation.
Mahmoud Abou Alaina (fig.1) is Assistant Professor of Internal Medicine-Pulmonary, Critical Care and Occupational Medicine, Department of Internal Medicine and Pappajohn Biomedical Institute, Roy J. and Lucille A. Carver College of Medicine, University of Iowa, Iowa City, Iowa, USA.
Adjemian J, Olivier KN, Prevots DR.Epidemiology of Pulmonary Nontuberculous Mycobacterial Sputum Positivity in Patients with Cystic Fibrosis in the United States, 2010-2014. Ann Am Thorac Soc. 2018 Jun 13. doi: 10.1513/AnnalsATS.201709-727OC. [Epub ahead of print] [Pubmed]

Fig. 2 Jennifer Adjeman
LinkedIn
Pulmonary nontuberculous mycobacterial (NTM) disease represents a significant threat to cystic fibrosis (CF) patients, with an estimated annual prevalence of 12%. Prior studies report an increasing annual NTM prevalence in the general population, though similar trends in persons with CF have not been assessed. This study aimed to identify the prevalence, geographic patterns, temporal trends and risk factors for NTM positivity by mycobacterial species among persons with CF throughout the United States using annualized CF Patient Registry (CFPR) data from 2010-2014. Of 16,153 included persons with CF, 3,211 (20%) had a pathogenic NTM species isolated at least once over the 5-year period; 1,949 (61%) had Mycobacterium avium complex (MAC) and 1,249 (39%) M. abscessus. NTM prevalence showed a significant relative increase of 5% per year, from 11.0% in 2010 to 13.4% in 2014 (p=0.0008), although this varied by geographic area. Study participants with either MAC or M. abscessus were significantly more likely to have been diagnosed with CF at an older age (p<0.0001), have a lower BMI (p<0.0001), higher forced expiratory volume in one second (FEV1) percent predicted (p<0.01), and fewer years on chronic macrolide therapy (p<0.0001).
The authors concluded NTM remains highly prevalent among adults and children with CF in the U.S., with one in five affected, and appears to be increasing over time. Variation in prevalence exists by geographic region and by patient-level factors, including older age and receiving an initial CF diagnosis later in life. Routine screening for NTM, including mycobacterial speciation, especially in high-risk geographic areas, is critical for better understanding its epidemiology and changes in prevalence over time.
Jennifer Adjemian (fig. 2) is at the Epidemiology Unit, Laboratory of Clinical Immunology and Microbiology, Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, Maryland.
Anand S, Mande SS. Diet, Microbiota and Gut-Lung Connection. Front Microbiol. 2018 Sep 19;9:2147. doi: 10.3389/fmicb.2018.02147. eCollection 2018. Free PMC Article [Pubmed]

Fig. 3 Swadha Anand
brics-ysf.org
The gut microbial community (Gut microbiota) is known to impact metabolic functions as well as immune responses in our body. Diet plays an important role in determining the composition of the gut microbiota. Gut microbes help in assimilating dietary nutrients which are indigestible by humans. The metabolites produced by them not only modulate gastro-intestinal immunity, but also impact distal organs like lung and brain. Micro-aspiration of gut bacteria or movement of sensitized immune cells through lymph or bloodstream can also influence immune response of other organs. Dysbiosis in gut microbiota has been implicated in several lung diseases, including allergy, asthma and cystic fibrosis. The bi-directional cross-talk between gut and lung (termed as Gut-Lung axis) is best exemplified by intestinal disturbances observed in lung diseases. Some of the existing probiotics show beneficial effects on lung health. A deeper understanding of the gut microbiome which comprises of all the genetic material within the gut microbiota and its role in respiratory disorders is likely to help in designing appropriate probiotic cocktails for therapeutic applications.
Swadha Anand (fig.3) is at Bio-Sciences R&D Division, TCS Research, Tata Consultancy Services Ltd., Pune, India.
Arooj P, Plant R, Doddakula K, Plant BJ. Inhaler screw-up: A real world cystic fibrosis (CF) case. Pediatr Pulmonol. 2018 Sep 18. doi: 10.1002/ppul.24158. [Epub ahead of print] [Pubmed]
-

Fig. 4 Parniya Arooj
- A 40-year-old male with Cystic Fibrosis developed a sudden onset choking, cough, dyspnea and hemoptysis after using his inhaler. He presented to A&E. CT scan located a foreign body in his right bronchus intermedius (RBI). Rigid bronchoscopy was performed and removed a plastic raw plug with screw from his RBI. He made a rapid recovery. On questioning he mentioned he had stored his inhaler uncapped in the same pocket he had this screw that he found at work.
The authors note that small objects can lodge in inhalers due to their design/mouthpiece uncapping. This can result in endobronchial foreign body aspiration. This case highlights the importance of replacing inhaler cap when not in use.
Dr. Parniya Arooj (fig.4) is a member of the Department of Respiratory Medicine, Cork University Hospital, Ireland
Nikhil T Awatade, Sharon L Wong, Chris K Hewson, Laura K Fawcett, Anthony Kicic, Adam Jaffe, Shafagh A Waters Human Primary Epithelial Cell Models: Promising Tools in the Era of Cystic Fibrosis Personalized Medicine. Front Pharmacol 2018 Dec 7;9:1429.doi: 10.3389/fphar.2018.01429. eCollection 2018. Free PMC article (very detailed fully referenced) [Pubmed]

Fig. 5 Nikhil Tanaji Awatade
loop.frontiersin.org
Cystic fibrosis (CF) is an inherited disorder where individual disease etiology and response to therapeutic intervention is impacted by CF transmembrane regulator (CFTR) mutations and other genetic modifiers. CFTR regulates multiple mechanisms in a diverse range of epithelial tissues.
In this Review, we consolidate the latest updates in the development of primary epithelial cellular model systems relevant for CF. We discuss conventional two-dimensional (2-D) airway epithelial cell cultures, the backbone of in vitro cellular models to date, as well as improved expansion protocols to overcome finite supply of the cellular source. We highlight a range of strategies for establishment of three-dimensional (3-D) airway and intestinal organoid models and evaluate the limitations and potential improvements in each system, focusing on their application in CF. The in vitro CFTR functional assays in patient-derived organoids allow for preclinical pharmacotherapy screening to identify responsive patients. It is likely that organoids will be an invaluable preclinical tool to unravel disease mechanisms, design novel treatments, and enable clinicians to provide personalized management for patients with CF.
Nikhil T Awatade (fig. 5) is a Post-Doctoral Fellow in the Faculty of Medicine, School of Women’s and Children’s Health and the Molecular and Integrative Cystic Fibrosis Research Centre, School of Medical Sciences, Faculty of Medicine, University of New South Wales, SydneyAustralia.
Ayoub F, Trillo-Alvarez C, Morelli G, Lascano J. Risk factors for hepatic steatosis in adults with cystic fibrosis: Similarities to non-alcoholic fatty liver disease. World J Hepatol. 2018 Jan 27;10(1):34-40. doi: 10.4254/wjh.v10.i1.34. [Pubmed]

Fig. 6 Fares W Ayoub
cdn.bcm.edu
To investigate the clinical, biochemical and imaging characteristics of adult cystic fibrosis (CF) patients with hepatic steatosis as compared to normal CF controls. Data was collected on 114 patients meeting inclusion criteria. Seventeen patients (14.9%) were found to have hepatic steatosis on imaging. Being overweight (BMI > 25) (P = 0.019) and having a higher ppFEV1 (75 vs 53, P = 0.037) were significantly associated with hepatic steatosis. Patients with hepatic steatosis had a significantly higher median alanine aminotransferase level (27 vs 19, P = 0.048). None of the hepatic steatosis patients had frank CF liver disease, cirrhosis or portal hypertension. We found no significant association with pancreatic insufficiency or CF related diabetes.
- The authors concluded hepatic steatosis appears to be a clinically and phenotypically distinct entity from CF liver disease. The lack of association with malnourishment and the significant association with higher BMI and higher ppFEV1 demonstrate similarities with non-alcoholic fatty liver disease. They suggest ong-term prospective studies are needed to ascertain whether CF hepatic steatosis progresses to fibrosis and cirrhosis.
Dr Fares Walid Ayoub (fig. 6) is a physician in the Department of Medicine, University of Florida, Gainsville, Florida. Subsequently Assistant Professor, Baylor College of Medicine.
Ballmann M, Hubert D, Assael BM, Staab D, Hebestreit A, Naehrlich L, Nickolay T, Prinz N, Holl RW; CFRD Study Group. Repaglinide versus insulin for newly diagnosed diabetes in patients with cystic fibrosis: a multicentre, open-label, randomised trial. Lancet . Endocrinol. 2018 Feb;6(2):114-121. doi: 10.1016/S2213-8587(17)30400-X. Epub 2017 Dec 5. [Pubmed]

Fig. 7 Marten Ballmann
medscape.org
A study to assess the efficacy and safety of oral anti-diabetic drugs. A multicentre, open-label, comparative, randomised trial in 49 centres in Austria, France, Germany, and Italy. Eligible patients had cystic fibrosis, were older than 10 years, and had newly diagnosed diabetes. Thirty-four patients were enrolled in the repaglinide group and 41 in the insulin group, of whom 30 and 37, respectively, were included in the analyses. At 24 months, glycaemic control was similar in the repaglinide and insulin groups (mean change in HbA1cconcentration from baseline 0·2% [SD 0·7%], 1·7 mmol/mol [8·1 mmol/mol] with repaglinide vs -0·2% [1·3%], -2·7 mmol/mol, [14·5 mmol/mol] with insulin; mean difference between groups -0·4%, (95% CI -1·1 to 0·2 [-4·4 mmol/mol, -11·5 to 2·7], p=0·15). The most frequent adverse events were pulmonary events (43 [40%] of 107 in the repaglinide group and 60 [45%] of 133 in the insulin group), and the most frequent serious adverse events were pulmonary events leading to hospital admission (five [50%] of ten and seven [54%] of 13, respectively.
The authors concluded that repaglinide for glycaemic control in patients with cystic-fibrosis-related diabetes is as efficacious and safe as insulin.
Manfred Ballmann (Fig. 7) is at the Paediatric Clinic, University Medicine Rostock, Rostock, Germany; Clinic for Paediatric Pulmonology, Allergy, and Neonatology, Medical School Hannover, Hannover, Germany.
Barnaby R, Koeppen K, Nymon A, Hampton TH, Berwin B, Ashare A, Stanton BA. Lumacaftor (VX-809) restores the ability of CF macrophages to phagocytose and kill Pseudomonas aeruginosa. Am J Physiol Lung Cell Mol Physiol. 2018 Mar 1;314(3):L432-L438. doi: 10.1152/ajplung.00461.2017. Epub 2017 Nov 16. [Pubmed]
Although ivacaftor (VX-770) alone and ivacaftor in combination with lumacaftor (VX-809) improve lung function in CF patients with the Gly551Asp and del508Phe mutations, respectively, the effects of these drugs on the function of human CF macrophages are unknown. Thus studies were conducted to examine the effects of lumacaftor alone and lumacaftor in combination with ivacaftor (i.e., ORKAMBI) on the ability of human CF ( del508Phe/ del508Phe) monocyte-derived macrophages (MDMs) to phagocytose and kill Pseudomonas aeruginosa.
Lumacaftor alone restored the ability of CF MDMs to phagocytose and kill P. aeruginosa to levels observed in MDMs obtained from non-CF (WT-CFTR) donors. This effect contrasts with the partial (~15%) correction of del508Phe Cl- secretion of airway epithelial cells by lumacaftor. Ivacaftor reduced the ability of lumacaftor to stimulate phagocytosis and killing of P. aeruginosa. Lumacaftor had no effect on P. aeruginosa-stimulated cytokine secretion by CF MDMs. Ivacaftor (5 µM) alone and ivacaftor in combination with lumacaftor reduced secretion of several proinflammatory cytokines. The clinical efficacy of ORKAMBI may be related in part to the ability of lumacaftor to stimulate phagocytosis and killing of P. aeruginosa by macrophages.
Roxanna Barnaby is in the Department of Microbiology and Immunology, Geisel School of Medicine at Dartmouth, Hanover, New Hampshire.
Berdah L, Taytard J, Leyronnas S, Clement A, Boelle PY, Corvol H. Stenotrophomonas maltophilia: A marker of lung disease severity Pediatr Pulmonol. 2018 Apr;53(4):426-430. doi: 10.1002/ppul.23943. Epub 2018 Jan 4. Free PMC pubmed.ncbi.nlm.nih.gov/29314745/
While the prevalence of Stenotrophomonas maltophilia lung infection in cystic fibrosis (CF) patients has increased in the last decades, its pathogenicity remains controversial. The aim of this study was to investigate the effects of S. maltophilia initial infection on the progression of lung disease in CF children. This case-control retrospective study took place in a pediatric CF centre. A total of 23 cases defined by at least one sputum culture positive for S. maltophilia, were matched for age, sex, and CFTR mutations to 23 never infected CF controls. The clinical data were collected for 2 years before and after S. maltophilia initial infection and comprised lung function analyses, rates of exacerbations and of antibiotic courses.
Compared with controls, cases had lower lung function (P = 0.05), more frequent pulmonary exacerbations (P = 0.01), hospitalizations (P = 0.02), and intravenous antibiotic courses (P = 0.04) before S. maltophilia acquisition. In the year following S. maltophilia initial infection, lung function decline was similar in cases and controls but cases remained more severe, with more frequent pulmonary exacerbations (P = 0.01), hospitalizations (P = 0.02) and intravenous antibiotics.
The authors concluded S. maltophilia seems to be a marker of CF lung disease severity and international recommendations to reduce lung infection by this pathogen should rapidly emerge (?)
Laura Berdah works at the APHP Hopital Trousseau, CF Center Paris.
Bessonova L, Volkova N, Higgins M, Bengtsson L, Tian S, Simard C, Konstan MW, Sawicki GS, Sewall A, Nyangoma S, Elbert A, Marshall BC, Bilton D.Data from the US and UK cystic fibrosis registries support disease modification by CFTR modulation with ivacaftor.Thorax. 2018 Aug;73(8):731-740. doi: 10.1136/thoraxjnl-2017-210394. Epub 2018 May 10. Free full text. [Pubmed]
-

Fig. 8 Leona Bessonova eMedEvents
- This on going, observational, post-approval safety study evaluates clinical outcomes and disease progression in ivacaftor-treated patients using data from the US and the UK CF registries following commercial availability. Results from the 2014 analyses (2 and 3 years following commercial availability in the UK and USA, respectively) are presented here.
Analyses included 1256 ivacaftor-treated and 6200 comparator patients from the USA and 411 ivacaftor-treated and 2069 comparator patients from the UK. No new safety concerns were identified based on the evaluation of clinical outcomes included in the analyses. As part of safety evaluations, ivacaftor-treated US patients were observed to have significantly lower risks of death (0.6% vs. 1.6%, p=0.0110), transplantation (0.2% vs. 1.1%, p=0.0017), hospitalisation (27.5% vs. 43.1%, p<0.0001) and pulmonary exacerbation (27.8% vs. 43.3%, p<0.0001) relative to comparators; trends were similar in the UK. In both registries, ivacaftor-treated patients had a lower prevalence of CF-related complications and select microorganisms and had better preserved lung function.
The authors concluded that while general limitations of observational research apply, analyses revealed favourable results for clinically important outcomes among ivacaftor-treated patients, adding to the growing body of literature supporting disease modification by CFTR modulation with ivacaftor.
Dr Leona Bessonova (fig.8) and Dr Nataliya Volkova are from Vertex Pharmaceuticals, Boston Massachusetts
Brandt C, Roehmel J, Rickerts V, Melichar V, Niemann N, Schwarz C. Aspergillus Bronchitis in Patients with Cystic Fibrosis. Mycopathologia. 2018 Feb;183(1):61-69. doi: 10.1007/s11046-017-0190-0. Epub 2017 Aug 17 [Pubmed]
- Aspergillus fumigatus frequently colonizes the airways of patients with cystic fibrosis (CF) and may cause various severe infections, such as bronchitis. Serological data, sputum dependent markers and longitudinal data of treated cases of Aspergillus bronchitis were evaluated for further description of this infection. This study, which comprises three substudies, aimed to analyze epidemiological data of Aspergillus in CF and the entity of Aspergillus bronchitis. A retrospective data analysis of 10 treated cases revealed the clinical course of Aspergillus bronchitis, including repeated positive sputum culture findings for A. fumigatus, no antibiotic treatment response, total serum IgE levels <200 kU/l, no observation of new pulmonary infiltrates and appropriate antifungal treatment response. Antifungal treatment durations of 4 ± 1.6 (2-6) weeks significantly reduced cough (P = 0.0067), sputum production (P < 0.0001) and lung function measures (P = 0.0358) but not physical capacity (P = 0.0794)
From this retrospective study, a prevalence of 1.6% was calculated. In addition, two cases of Aspergillus bronchitis were identified in the prospective cohort study according to immunological, molecular and microbiological parameters. A prevalence of 9% was assessed.
Aspergillus bronchitis appears to occur in a minority of colonized CF patients. Antifungal treatment may reduce respiratory symptoms and restore lung function.
Claudia Brandt is in the Department of Pediatric Pneumology and Immunology, Cystic Fibrosis Center, Charité-Universitätsmedizin Berlin, Augustenburger Platz 1, 13353, Berlin, Germany.
Bridges N, Rowe R, Holt RIG. Unique challenges of cystic fibrosis-related diabetes. Diabet Med. 2018 Apr 23. doi: 10.1111/dme.13652. [Epub ahead of print] [Pubmed]
Individuals with cystic fibrosis and pancreatic insufficiency have a gradual decline in insulin secretion over time, which results in an increase in the prevalence of diabetes with age; up to 50% of adults with cystic fibrosis aged over 35-40 years have diabetes. Cystic fibrosis-related diabetes differs from Type 1 and Type 2 diabetes in several ways; there is a pattern of insulin deficiency with reduced and delayed insulin response to carbohydrates but a sparing of basal insulin that results in glucose abnormalities, which are frequently characterized by normal fasting glucose and postprandial hyperglycaemia. Insulin deficiency and hyperglycaemia, even at levels which do not reach the threshold for a diagnosis of diabetes, have an adverse impact on lung function and clinical status in people with cystic fibrosis. Although the risk of microvascular complications occurs as in other forms of diabetes, the main reason for treatment is to prevent deterioration in lung function and weight loss; treatment may therefore be required at an earlier stage than for other types of diabetes. Treatment is usually with insulin, but management needs to take into account all the other medical issues that arise in cystic fibrosis.
-
-

- fig. 9 Richard Holt
-

- fig. 10 Rachel Rowe
-

- Fig.11 Nicola Bridges
-
- Professor Richard Holt (fig.9) is Professor in Diabetes and Endocrinology within Medicine at the University of Southampton
Dr. Nicola Bridges (fig.11) is a Paediatric Endocrinologist based at Chelsea and Westminster Hospital, London and also works at the Royal Brompton Hospital and St Mary’s Hospital. - Dr. Rachel Rowe (fig 10) is Consultant Physician, Diabetologist at the University Hospital of South Manchester NHS Foundation Trust.
Breuer O, Caudri D, Akesson L, Ranganathan S, Stick SM, Schultz A; AREST CF. The clinical significance of oropharyngeal cultures in young children with cystic fibrosis. 2018 Apr 20. pii: 1800238. doi: 10.1183/13993003.00238-2018. [Epub ahead of print] [Pubmed]

Fig. 12 Daan Caudri
ResearchGate

Fig.11 Oded Breuer Walyan Respiratory Research
In children with cystic fibrosis (CF) the associations between oropharyngeal swabs (OPS) for detection of Pseudomonas and lung disease has not been evaluated. OPS and bronchoalveolar lavage (BAL) samples were obtained annually in children with CF from 2005 to 2017. OPS test characteristics were calculated using BAL as gold standard. Results were related to lung inflammation (BAL neutrophil elastase, interleukin-8), structural lung disease (chest CT PRAGMA-CF scores), respiratory exacerbations, and future detection of Pseudomonas on BAL.
From 181 patients, 690 paired OPS-BAL cultures were obtained. Prevalence of Pseudomonas in BAL was 7.4%. OPS sensitivity was 23.0% and specificity 91.4%, reducing the post-test probability for a positive BAL following negative OPS to 6.3%. Pseudomonas on OPS was not associated with lung inflammation or respiratory exacerbations but was weakly associated with current PRAGMA-CF disease score (p=0.043). Pseudomonas on BAL was associated with positive neutrophil elastase (OR 4.17 CI95% 2.04-8.53, p<0.001), increased interleukin-8 (p<0.001), increased all baseline PRAGMA-CF scores (p<0.001), progression of PRAGMA-CF scores (p<0.05) and increased risk of respiratory exacerbations (IRR 2.11 CI95% 1.15-3.87, p=0.017).
In children with CF oropharyngeal swabs only marginally change the probability of detecting lower airway Pseudomonas and are not associated with lung disease indices nor exacerbations risk.
First Authors – Oded Breuer (fig. 11) is a paediatric pulmonologist at Telethon Kids Institute, University of Western Australia, Perth, Australia and Princess Margaret Hospital for Children, Perth, Australia.
Daan Caudri (fig. 12) is a paediatric pulmonologist at Telethon Kids Institute, University of Western Australia, Perth, Australia. Princess Margaret Hospital for Children, Perth, Australia and Dept of Pediatrics/Respiratory Medicine, Erasmus MC, Rotterdam, The Netherlands.
Bruch BA, Singh SB, Ramsey LJ, Starner TD. Impact of a cystic fibrosis transmembrane conductance regulator (CFTR) modulator on high-dose ibuprofen therapy in pediatric cystic fibrosis patients. Pediatr Pulmonol. 2018 Aug; 53(8):1935-1039. . doi: 10.1002/ppul.24024. [Epub ahead of print]May 1 [Pubmed]

Fig. 13 Brittany Bruch
University of Iowa
This study was undertaken to determine if a clinically relevant drug-drug interaction occurred between ibuprofen and lumacaftor/ivacaftor. Peak ibuprofen plasma concentrations were measured prior to and after lumacaftor/ivacaftor initiation. The authors showed a clinically relevant drug-drug interaction exists between ibuprofen and lumacaftor/ivacaftor. Lumacaftor may cause sub-therapeutic ibuprofen plasma concentrations due to the induction of CYP enzymes and increased metabolism of ibuprofen. Based on this analysis, they have modified their use of ibuprofen in several patients after evaluation of this drug-drug interaction.
Brittany Bruch (fig.13) is a pharmacist with University of Iowa Hospitals and Clinics, Iowa City, Iowa.
Byrnes LJ, Xu Y, Qiu X, Hall JD, West GM.Sites associated with Kalydeco binding on human Cystic Fibrosis Transmembrane Conductance Regulator revealed by Hydrogen/Deuterium Exchange. Sci Rep. 2018 Mar 16;8(1):4664. doi: 10.1038/s41598-018-22959-6. Free PMC Article [Pubmed]
-

Fig. 14 Laura Jean Byrnes
- Cystic Fibrosis (CF) is caused by mutations in the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR). Mutations associated with CF cause loss-of-function in CFTR leading to salt imbalance in epithelial tissues. Kalydeco (also called VX-770 or ivacaftor) was approved for CF treatment in 2012 but little is known regarding the compound’s interactions with CFTR including the site of binding or mechanisms of action. In this study we use hydrogen/deuterium exchange (HDX) coupled with mass spectrometry to assess the conformational dynamics of a thermostabilized form of CFTR in apo and ligand-bound states. We observe HDX protection at a known binding site for AMPPNP and significant protection for several regions of CFTR in the presence of Kalydeco. The ligand-induced changes of CFTR in the presence of Kalydeco suggest a potential binding site.
Dr. Laura Jean Byrnes (fig.14) is a scientist at Pfizer, Cornell University
Carlile GW, Yang Q, Matthes E, Liao J, Radinovic S, Miyamoto C, Robert R, Hanrahan JW, Thomas DY.A novel triple combination of pharmacological chaperones improves F508del-CFTR correction. Sci Rep. 2018 Jul 30;8(1):11404. doi: 10.1038/s41598-018-29276-y Free PMC Article [Pubmed]

Fig 15 Graeme Carlile
ResearchGate
Pharmacological chaperones (e.g. VX-809, lumacaftor) that bind directly to F508del-CFTR and correct its mislocalization are promising therapeutics for Cystic Fibrosis (CF). However to date, individual correctors provide only ~4% improvement in lung function measured as FEV1, suggesting that multiple drugs will be needed to achieve substantial clinical benefit.
Here we examine if multiple sites for pharmacological chaperones exist and can be targeted to enhance the rescue of F508del-CFTR with the premise that additive or synergistic rescue by multiple pharmacological chaperones compared to single correctors indicates that they have different sites of action.
First, we found that a combination of the pharmacological chaperones VX-809 and RDR1 provide additive correction of F508del-CFTR. Then using cellular thermal stability assays (CETSA) we demonstrated the possibility of a third pharmacologically important site using the novel pharmacological chaperone tool compound 4-methyl-N-[3-(morpholin-4-yl) quinoxalin-2-yl] benzenesulfonamide (MCG1516A). All three pharmacological chaperones appear to interact with the first nucleotide-binding domain (NBD1). The triple combination of MCG1516A, RDR1, and VX-809 restored CFTR function to >20% that of non-CF cells in well differentiated HBE cells and to much higher levels in other cell types. Thus the results suggest the presence of at least three distinct sites for pharmacological chaperones on F508del-CFTR NBD1, encouraging the development of triple corrector combinations.
Graeme W Carlile (fig.15) is Research Associate at the Cystic Fibrosis Translational Research Center, Department of Biochemistry McGill University Montreal Quebec Canada, H3G 1Y6, Montreal, Quebec, Canada.
Carr SB, Ronan P, Lorenc A, Mian A, Madge SL, Robinson N.Children and Adults Tai Chi Study (CF-CATS2): a randomised controlled feasibility study comparing internet-delivered with face-to-face Tai Chi lessons in cystic fibrosis. ERJ Open Res. 2018 Dec 14;4(4). pii: 00042-2018. doi: 10.1183/23120541.00042-2018. eCollection 2018 Oct. 30568967 [Pubmed]
-

Fig. 16 Siobhan B Carr Royal Brompton Hospital
This study aimed to assess the feasibility, safety and acceptability of learning Tai Chi via an internet-based approach and compared patient-reported outcomes. Children and adults with cystic fibrosis were recruited to a randomised, comparative effectiveness trial. Participants learnt eight Tai Chi movements; teaching was delivered in eight lessons over 3 months: delivered either via the internet or face-to-face. Assessments were at 3-monthly intervals over 9 months. Outcomes included health status, quality of life, sleep, mindfulness and instructor-led questions. 40 adults and children completed the eight sets of Tai Chi lessons. The median age was 22.8 years (range 6.1-51.5 years); 27 patients were female. The cohort comprised 26 adults (aged >16 years), six teenagers and eight children (aged <12 years). The groups were well matched. Feasibility and safety were demonstrated. Participants showed significant improvements in self-reported sleep, cough (both daytime and night-time), stomach ache and breathing. No differences in lung function, health status, quality of life, sleep or mindfulness was shown before or after completing the lessons. Tai Chi was safe and well tolerated; it was feasible to deliver individual lessons via the internet, reducing concerns regarding cross- infection, and appeared to improve self-reported symptoms.
Dr Siobhan B Carr (fig.16) is consultant in paediatric respiratory medicine with a special interest in suppurative lung disease at the Royal Brompton Hospital London. She is also Chair of the UK CF Registry Steering Committe
Carrion A, Borowitz DS, Freedman SD, Siracusa CM, Goralski JL, Hadjiliadis D, Srinivasan S, Stokes DC. Reduction of Recurrence Risk of Pancreatitis in Cystic Fibrosis With Ivacaftor: Case Series. J Pediatr Gastroenterol Nutr. 2018 Mar;66(3):451-454. doi: 10.1097/MPG.0000000000001788. pubmed.ncbi.nlm.nih.gov/29045347/
-

Fig.17 Andres Carrion Find a Doctor
A multi center retrospective study of patients with CF taking ivacaftor who had a history of recurrent pancreatitis. During the first 3 months of therapy, only 1 of the 6 patients had an episode of pancreatitis, which was managed on an outpatient basis. Between 3 and 12 months on ivacaftor therapy, none of the patients had recurrence of pancreatitis or required hospitalization. The use of ivacaftor was associated with a reduced frequency and recurrence rate of pancreatitis in patients with CF.
Dr Andres Carrion (fig.17) specialises in Gastroenterology and Transplant Hepatology in the University Miami Health.
Caskey S, Stirling J, Moore JE, Rendall JC. Occurrence of Pseudomonas aeruginosa in waters: Implications for patients with cystic fibrosis(CF). Lett Appl Microbiol. 2018 Mar 14. doi: 10.1111/lam.12876. [Epub ahead of print] pubmed.ncbi.nlm.nih.gov/29537700/

Fig. 18 Steven Caskey
ResearchGate
Chronic P. aeruginosa infection is associated with increased morbidity and mortality in patients with cystic fibrosis (CF). Current understanding of risk factors for acquisition is limited and so the aim of this study was to examine a large sample of environmental waters from diverse sources. Environmental water samples [n= 7904] from jacuzzis, hydrants, swimming pools, hot tubs, plunge pools, bottled natural mineral water (NMW), taps, springs, ice machines, water coolers, bores and showers were examined for the presence of P. aeruginosa.
P. aeruginosa was detected in 524/7904 (6.6%) waters examined. Hot tubs [51/243; 20.9%], tap water [3/40; 8%] and jacuzzis [432/5811; 7.4%] were the most likely environments where P. aeruginosa was isolated. P. aeruginosa was isolated from bottled water [2/67; 3%].
The authors say their study highlights the ubiquitous nature of P. aeruginosa in the environment. Given CF patients are frequently counselled to make lifestyle changes to minimize P. aeruginosa exposure, these results have important implications. In particular, the occurrence of P. aeruginosa in tap water highlights the need to disinfect the CF patients’ nebuliser after each use.
— A study from John Moore’s unit in Belfast confirming the ubiquitous nature of P. aeruginosa in the environment.Confirming the particular dangers of hot tubs.
Steven Casey (fig.18) is a consultant in cystic fibrosis at the Regional Adult Cystic Fibrosis Centre, Level 8, Belfast City Hospital, Belfast, UK.
Castellani C, Duff AJA, Bell SC, Heijerman HGM, Munck A, Ratjen F, Sermet-Gaudelus I, Southern KW, Barben J, Flume PA, Hodková P, Kashirskaya N, Kirszenbaum MN, Madge S, Oxley H, Plant B, Schwarzenberg SJ, Smyth AR, Taccetti G, Wagner TOF, Wolfe SP, Drevinek P.ECFS best practice guidelines: the 2018 revision. J Cyst Fibros. 2018 Mar;17(2):153-178. doi: 10.1016/j.jcf.2018.02.006. Epub 2018 Mar 3. [Pubmed] Free full text
Developments in managing CF continue to drive dramatic improvements in survival. As newborn screening rolls-out across Europe, CF centres are increasingly caring for cohorts of patients who have minimal lung disease on diagnosis. With the introduction of mutation-specific therapies and the prospect of truly personalised medicine, patients have the potential to enjoy good quality of life in adulthood with ever-increasing life expectancy.
The landmark Standards of Care published in 2005 set out what high quality CF care is and how it can be delivered throughout Europe. This underwent a fundamental re-write in 2014, resulting in three documents; center framework, quality management and best practice guidelines. This document is a revision of the latter, updating standards for best practice in key aspects of CF care, in the context of a fast-moving and dynamic field. In continuing to give a broad overview of the standards expected for newborn screening, diagnosis, preventative treatment of lung disease, nutrition, complications, transplant/end of life care and psychological support, this consensus on best practice is expected to prove useful to clinical teams both in countries where CF care is developing and those with established CF centres. The document is an ECFS product and endorsed by the CF Network in ERN LUNG and CF Europe.
Castellani C, Boner AL.Aquagenic wrinkling and cystic fibrosis carriership: A dubious relationship. Eur J Intern Med. 2018 Sep 19. pii: S0953-6205(18)30370-4. doi: 10.1016/j.ejim.2018.09.007. [Epub ahead of print] [Pubmed]
-

Fig. 19 Carlo Castellani
- There are 32 references to aquagenic wrinkling since 2004. The first report was by Prof. Bob Elliot from New Zealand in 1974 (see Seventies Clinical section of this History); the first report caused great excitement at the time. The present authors are concerned that AW has been suggested as a manifestation of CF carriership; they suggest there are three arguments against this.
First, the high carrier frequency in Europeans would make AW a fairly common complaint. Second, CF carriers are asymptomatic and there is no reason for AW to be an exception. Third, a case report of an individual F508del/G551D treated with ivacaftor showed resolution of AW (Bielicky L et al. 2015. Abstract in 2015 section) Although anecdotal, the evidence of AW disappearance following the rescue of CFTR function in one of the two affected alleles suggests that a single mutated allele is not sufficient to determine AW.
The authors suggest that that some of these supposed carriers are actually compound heterozygotes and carry another, undetected, sequence variation in the gene.
Dr. Carlo Castellani (fig.19) Is based at the Cystic Fibrosis Centre, Istituto Gaslini, Genoa, Italy. He received the ECFS Award for his extensive work in cystic fibrosis.
Cho DY, Zhang S, Lazrak A Grayson JW, Peña Garcia JA, Skinner DF, Lim DJ, Mackey C, Banks C, Matalon S, Woodworth BA. Resveratrol and ivacaftor are additive G551D CFTR-channel potentiators: therapeutic implications for cystic fibrosis sinus disease. Int Forum Allergy Rhinol. 2018 Aug 27. doi: 10.1002/alr.22202. [Epub ahead of print][Pubmed]
-

Fig.20 Do-Yeon Cho Scholars.uab.edu
- Ivacaftor is a CFTR potentiator that improves Cl- transport in CF patients with at least 1 copy of the G551D mutation. Resveratrol is also a potent CFTR potentiator that increases determinants of mucociliary transport. The objective of this study is to determine whether resveratrol and ivacaftor improve Cl- secretion in G551D CFTR over either agent alone. G551D Fisher rat thyroid cells there was improvement in G551D CFTR-mediated Cl- secretion suggests that resveratrol could enhance ivacaftor therapy in these patients and improve CF-related rhino sinusitis.
Dr. Do-Yeon Cho (fig 20) is Director of Otolaryngology Research, Department of Otolaryngology, University of Alabama, Birmingham Alabama
Chokoshvili D, Vears D, Borry P. Expanded carrier screening for monogenic disorders: where are we now? Prenat Diagn. 2018 Jan;38(1):59-66. doi: 10.1002/pd.5109. Epub 2017 Jul 27 [Pubmed]
-

Fig. 21. Davit Chokoshvili
- To identify relevant expanded carrier screening (ECS) providers, we employed a multi-step approach, which included online searching, review of the recent literature, and consultations with researchers familiar with the current landscape of ECS. As of January 2017, there were 16 providers of ECS tests: 13 commercial companies, 2 medical hospitals, and 1 academic diagnostic laboratory. The authors noted drastic differences in the characteristics of ECS tests, with the number of conditions ranging from 41 to 1792. Only three conditions (cystic fibrosis, maple syrup urine disease 1b, and Niemann-Pick disease) were screened for by all providers. Where the same disease gene was included by multiple providers, substantial differences existed in the mutations screened and/or variant interpretation/reporting strategie
The authors suggested that, given the importance of carrier screening results in reproductive decision-making, the observed heterogeneity across ECS panels is concerning. Efforts should be made to ensure that clear and concrete criteria are in place to guide the development of ECS panels.
Dr. Davit Chokoshvili (fig.21) is a doctoral researcher at the Centre for Biomedical Ethics and Law. Leuven
Cirilli N, Raia V, Rocco I, De Gregorio F, Tosco A, Salvadori L, Sepe AO, Buzzetti R, Minicuci N, Castaldo G.Intra-individual biological variation in sweat chloride concentrations in CF, CFTR dysfunction, and healthy pediatric subjects.Pediatr Pulmonol. 2018 Apr 2. doi: 10.1002/ppul.23992. [Epub ahead of print] [Pubmed]
-

Fig. 22 Natalia Cirilli fibrosicisticaricerca.it
- The sweat test is one of the main diagnostic tools used in newborn screening programs and as a confirmatory test, in case of suspect of Cystic Fibrosis (CF). Since sweat chloride (Cl) concentration is also considered an appropriate parameter to explore the efficacy of CFTR modulators in clinical trials, it is crucial to evaluate the biological variability of this test in healthy and pathological conditions. The aim of this pilot study was to determine the individual biological variability of sweat Cl, both in healthy individuals and CF patients and to assess its correlation with diet, season, and menstrual cycle. Thirty-five out of 36 selected subjects (6-18 years) were enrolled by 2 CF care centers and assigned to 3 cohorts: CF, CFTR-related disorder (CFTR-RD) and healthy volunteers. Each participant was subjected to eight sweat tests in different conditions and time of the year. Data were analyzed using linear mixed effects models for repeated measures, taking also into account intra-individual correlations.
The authors observed a high intra-individual variability of sweat Cl, with the lowest mean CV% values among CF patients (20.21 in CF, 29.74 in CFTR-RD, and 31.15 in healthy subjects). Gender and diet had no influence on sweat Cl variability, nor had pubertal age and menstrual phase.
Results of this pilot study confirmed that sweat Cl variability is high in CF patients, although non-CF individuals displayed even higher mean CV% values. Season significantly influenced sweat test values only in CF patients, likely due to changes in their hydration status.
Dr. Natalia Cirilli (fig.22) is a scientist at the Mother Child Department, CF Referral Centre, Ancona, Italy
— The sweat test remains an important investigation even 65 years after the sweat electrolyte abnormality was first discovered by Paul Sant’Agnese and Robert Darling, although the CV%s do appear to limit its value as a measure of efficacy of the newer corrective treatments.
Clancy JP, Cotton CU, Donaldson SH, Solomon GM, VanDevanter DR, Boyle MP, Gentzsch M, Nick JA, Illek B, Wallenburg JC, Sorscher EJ, Amaral MD, Beekman JM, Naren AP, Bridges RJ, Thomas PJ, Cutting G, Rowe S, Durmowicz AG, Mense M, Boeck KD, Skach W, Penland C, Joseloff E, Bihler H, Mahoney J, Borowitz D, Tuggle KL. CFTR modulator theratyping: Current status, gaps and future directions. J Cyst Fibros. 2018 Jun 19. pii: S1569-1993(18)30585-X. doi: 10.1016/j.jcf.2018.05.004. [Epub ahead of print] Free full text [Pubmed]
The Cystic Fibrosis Foundation (CFF) assembled a workshop of international experts to discuss the use of preclinical model systems to examine the nature of CF-causing variants in CFTR and the role of in vitro CFTR modulator testing to inform in vivo modulator use. The theme of the workshop was centred on CFTR theratyping, a term that encompasses the use of CFTR modulators to define defects in CFTR in vitro, with application to both common and rare CFTR variants.
Several preclinical model systems were identified in various stages of maturity, ranging from the expression of CFTR variant cDNA in stable cell lines to examination of cells derived from CF patients, including the gastrointestinal tract, the respiratory tree, and the blood. Common themes included the on going need for standardization, validation, and defining the predictive capacity of data derived from model systems to estimate clinical outcomes from modulator-treated CF patients.
The authors concluded CFTR modulator theratyping is a novel and rapidly evolving field that has the potential to identify rare CFTR variants that are responsive to approved drugs or drugs in development
Clancy JP. Rapid therapeutic advances in CFTR modulator science. Pediatr Pulmonol. 2018 Nov;53(S3):S4-S11. doi: 10.1002/ppul.24157.[Pubmed]

Fig. 23 John Clancy
Established CF therapies treat the downstream consequences of CFTR dysfunction and have led to steady improvements in patient survival. A class of drugs termed CFTR modulators has recently entered the CF therapeutic landscape. These drugs differ fundamentally from prior therapies in that they aim to improve the function of disease-causing CFTR variants. This review summarizes the science behind CFTR modulators, including their targets, mechanism of action, clinical benefit, and future directions in the field. CFTR modulators have dramatically changed how CF is treated, validated CFTR as a therapeutic target, and opened the door to truly personalized therapies and treatment regim
Dr. John Clancy (fig. 23) from the Department of Pediatrics, Cincinnati Children’s Hospital Medical Center, Cincinnati, Ohio.
Collins S. Nutritional management of cystic fibrosis – an update for the 21stcentury. Paediatr Respir Rev. 2018; 26:4-6. [Pubmed]

Fig. 24 Sarah Collins
Nutritional management is an essential part of multidisciplinary care for infants, children and adults with cystic fibrosis (CF). In 2016 two updated nutritional consensus guidelines were published. This review explores some of the key points in the nutritional management of people with CF in the 21st Century.
Sarah Collins (fig.24) is HEE/NIHR Clinical Doctoral Research Fellow/CF Specialist Dietitian, Royal Brompton and Harefield NHS Foundation Trust
— A concise up to date guidance on current best practice for nutritional management.
Cortes-Santiago N, Leung DH, Castro E, Finegold M, Wu H, Patel K. Hepatic Steatosis is Prevalent following Orthotopic Liver Transplantation in Children with Cystic Fibrosis. J Pediatr Gastroenterol Nutr. 2018 Sep 18. doi: 10.1097/MPG.0000000000002154. [Epub ahead of print] [Pubmed]

Fig. 25 Nadir Cortez-Santiago Baylor College of Medicine
A retrospective clinic-pathologic review of explants and allograft liver biopsies from 13 children and adolescents with CFLD. In this study, the median age at LT for CFLD was 15.7 years. Notably, 10 of 13 (77%) CF explants had >5% steatosis and 8 of 13 (61.5%) demonstrated variable fibrosis. The median age, gender, type of transplant (liver vs liver-lung), pancreatic insufficiency (PI) status, BMI%ile, genotype and prevalence of diabetes were comparable in those with and without explant steatosis. More than half of allograft biopsies showed significant steatosis (17/31, 54.8%) and lobular inflammation (16/31, 51.6%). Hepatocyte ballooning was less frequent (5/31, 16.1%). Overall, 6 patients (46.2%) had allograft steatosis that worsened over time in 2 patients (33%). None had advanced fibrosis (≥stage 3). Patients with allograft steatosis had significantly more biopsies, were more likely to be ‘liver only’ recipients, had a shorter interval since transplant and higher BMI%ile (though <85). Patients without explant steatosis never demonstrated allograft steatosis, while 60% of patients with explant steatosis (n = 6) developed varying degrees of allograft steatosis. The degree of explant steatosis did not predict its severity in allografts (p = 0.3)
— This is the first study highlighting the development of allograft steatosis in CF patients. The findings suggest that allograft steatosis in patients with CF may be related to pre-existing steatosis in native livers, regardless of other risk factors and may have implications on patient management and long-term graft/patient survival.
Dr. Nahir Cortes-Santiago (fig.25) is resident pathologist at Baylor College of Medicine. Texas
Davies JC, Moskowitz SM, Brown C, Horsley A, Mall MA, McKone EF, Plant BJ, Prais D, Ramsey BW, Taylor-Cousar JL, Tullis E, Uluer A, McKee CM, Robertson S, Shilling RA, Simard C, Van Goor F, Waltz D, Xuan F, Young T, Rowe SM; VX16-659-101 Study Group.VX-659-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles.N Engl J Med. 2018 Oct 25;379(17):1599-1611. doi: 10.1056/NEJMoa1807119. Epub 2018 Oct 18. 30334693?? [Pubmed]

Fig. 26 Jane Davies imperial.ac.uk
The next-generation cystic fibrosis transmembrane conductance regulator (CFTR) corrector VX-659, in triple combination with tezacaftor and ivacaftor (VX-659-tezacaftor-ivacaftor), was developed to restore the function of Phe508del CFTR protein in patients with cystic fibrosis.The effects of VX-659-tezacaftor-ivacaftor on the processing, trafficking, and function of Phe508del CFTR protein were investigated using human bronchial epithelial cells. A range of oral VX-659-tezacaftor-ivacaftor doses in triple combination were then evaluated in randomized, controlled, double-blind, multicenter trials involving patients with cystic fibrosis who were heterozygous for the Phe508del CFTR mutation and a minimal-function CFTR mutation (Phe508del-MF genotypes) or homozygous for the Phe508del CFTR mutation (Phe508del-Phe508del genotype). The primary end points were safety and the absolute change from baseline in the percentage of predicted forced expiratory volume in 1 second (FEV1).
RESULTS:(Trial numbers NCT03224351 and NCT03029455) VX-659-tezacaftor-ivacaftor significantly improved the processing and trafficking of Phe508del CFTR protein as well as chloride transport in vitro. In patients, VX-659-tezacaftor-ivacaftor had an acceptable safety and side-effect profile. Most adverse events were mild or moderate.
VX-659-tezacaftor-ivacaftor resulted in significant mean increases in the percentage of predicted FEV1 through day 29 (P<0.001) of up to 13.3 points in patients with Phe508del-MF genotypes; in patients with the Phe508del-Phe508del genotype already receiving tezacaftor-ivacaftor, adding VX-659 resulted in a further 9.7-point increase in the percentage of predicted FEV1. The sweat chloride concentrations and scores on the respiratory domain of the Cystic Fibrosis Questionnaire-Revised improved in both patient populations.
Jane Davies (fig.26) is professor at Imperial College London and Royal Brompton and Harefield NHS Foundation Trust, London (J.C.D.),
The authors (all listed in the PubMed link) concluded robust in vitro activity of VX-659-tezacaftor-ivacaftor targeting Phe508del CFTR protein translated into improvements for patients with Phe508del-MF or Phe508del-Phe508del genotypes. VX-659 triple-combination regimens have the potential to treat the underlying cause of disease in approximately 90% of patients with cystic fibrosis.
Desai CS, Vonderau JS, McCall R, Khan KM, Baron TH. Pancreatic cystosis in patients with cystic fibrosis: A qualitative systematic review. Pancreatology. 2018 Oct;18(7):700-704. doi: 10.1016/j.pan.2018.08.008. Epub 2018 Aug 17.[Pubmed]

Fig 27 Chirag Desai med.unc.edu
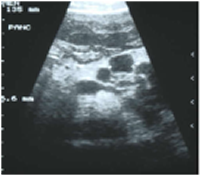
Fig. 28 Pancreatic cysts in a teenage boy with CF
Patients with cystic fibrosis (CF) and a CFTR gene mutation may present with a variety of pancreatic disorders. The presence of multiple macrocysts (>1 cm) replacing the entire pancreatic parenchyma is termed pancreatic cystosis. Lack of clear data makes clinical decision making challenging and controversial. The aim of this review is to perform a qualitative systematic analysis of the literature with intention to evaluate management plans.
The data of 24 patients were collected from included studies. Eight cases (33%) had a documented CFTR gene mutation and 10 (42%) were symptomatic at presentation. Imaging modalities included ultrasound in 18 (75%), CT in 12 (50%), and MRI in 8 (33%) cases. An average size of the largest cyst was 5.4 cm. 6 (25%) patients were offered therapy that described surgical (3), endoscopic (1), or medical therapy (2). Surgeries offered included total pancreatectomy, partial pancreatic resection of uncertain extent, and complex cyst resection. Endoscopic treatment was cystogastrostomy. Novel medical treatment was utilized with Doxepin, Propantheline, and Clonidine, resulting in reduction in cyst size and overall clinical improvement.
The authors suggest patients with pancreatic cystosis should not be denied treatment when necessary. This literature review is the most comprehensive thus far of cystic fibrosis and pancreatic cystosis. It did not provide identification of a definitive treatment plan or demonstrate contraindication to specific therapies
Chirag Desai (fig.28) is Professor in the Department of Surgery, University of North Carolina, USA
Donaldson SH, Pilewski JM, Griese M, Cooke J, Viswanathan L, Tullis E, Davies JC, Lekstrom-Himes JA, Wang LT5; VX11-661-101 Study Group. Tezacaftor/Ivacaftor in Subjects with Cystic Fibrosis and F508del/F508del-CFTR or F508del/G551D-CFTR. Am J Respir Crit Care Med. 2018 Jan 15;197(2):214-224. doi: 10.1164/rccm.201704-0717OC. [Pubmed]

Fig. 29 Scott H Donaldson
A study to evaluate the safety and efficacy of tezacaftor (formerly VX-661) monotherapy and of tezacaftor/ivacaftor combination therapy in subjects with cystic fibrosis homozygous for F508del or compound heterozygous for F508del and G551D. This was a randomized, placebo-controlled, double-blind, multicenter, phase 2 study. Subjects homozygous for F508del received tezacaftor (10 to 150 mg) every day alone or in combination with ivacaftor (150 mg every 12 h) in a dose escalation phase, as well as in a dosage regimen testing phase. Subjects compound heterozygous for F508del and G551D, taking physician-prescribed ivacaftor, received tezacaftor (100 mg every day).
Primary endpoints were safety through Day 56 and change in sweat chloride from baseline through Day 28. Secondary endpoints included change in per cent predicted FEV1 (ppFEV1) from baseline through Day 28 and pharmacokinetics. The incidence of adverse events was similar across treatment arms. Tezacaftor (100 mg every day)/ivacaftor (150 mg every 12 h) resulted in a 6.04 mmol/L decrease in sweat chloride and 3.75 percentage point increase in ppFEV1 in subjects homozygous for F508del, and a 7.02 mmol/L decrease in sweat chloride and 4.60 percentage point increase in ppFEV1 in subjects compound heterozygous for F508del and G551D from baseline through Day 28 (P < 0.05 for all).
The authors considered these results support continued clinical development of tezacaftor (100 mg every day) in combination with ivacaftor (150 mg every 12 h) in subjects with cystic fibrosis.
Scott Donaldson (fig. 29) is a pulmonologist and professor in the University of North Carolina School of Medicine, Chapel Hill, North Carolina.
Di Pietro ML, Teleman AA, Gonzalez-Melado FJ, Zace D, Di Raimo FR, Lucidi V, Refolo P. Implementing carrier screening for cystic fibrosis outside the clinic: ethical analysis in the light of the personalist view. 2018 Mar-Apr;169(2):e71-e76. doi: 10.7417/T.2018.2057. Free full text 29595869 [Pubmed]

Fig. 30 Pietro Refolo publires.unicatt.it
Cystic Fibrosis (CF) is an autosomal recessive genetic disease. Two models for screening CF are normally used: newborn screening and population-based CF carrier screening. In turn, there are three main models of population-based CF carrier screening: prenatal carrier screening, preconception carrier screening, and carrier screening outside clinical settings. Purpose of this study was to evaluate, in the light of the personalist view, the use of carrier screenings for CF outside the clinic, i.e. in non-clinical settings, such as school and workplaces.
An analysis has been carried out according to the “Personalist approach” (also called “Triangular model”), an ethical method for performing ethical analysis within HTA process. It includes factual, anthropological and ethical data in a ”triangular” normative reflection process.
Implementing carrier screening for cystic fibrosis outside the clinical settings allows acquisition of knowledge for informing reproductive choices, that can be considered as valuable; benefit-risk ratio seems to be not much favourable; autonomous and responsible decisions can be taken only under certain conditions; economic advantage is difficult to determine; therefore, from a personalist view, implementing carrier screenings outside the clinic seems not to be ethically justified.
So these authors conclude in accordance with the personalist perspective, public health programs providing carrier screenings outside the clinic should not be implemented.
Corresponding author is Dr. Pietro Refolo (fig.30) is associate professor at the Institute of Bioethics and Medical Humanities, Università Cattolica del Sacro Cuore,Fondazione Policlinico Universitario Agostino Gemelli, Rome, Italy.
— This article is difficult for anyone not already familiar with “personalism” (including the present writer!). Described as a theory or system based on subjective ideas or applications. The theory that probabilities do not have objective meaning but are expressions of a personal perspective on the occurrence of events. A system of thought which maintains the primacy of the human or divine person on the basis that reality has meaning only through the conscious mind.
Certainly the authors emphasise some of the real problems such as the screened person’s lack of understanding of the meaning of the results. Yet one cannot agree with their conclusions. This writer (JML) believes the CF is now largely a preventable condition and both carrier screening and also in preimplantation genetic diagnosis should be more widely publicised and be available to all.
Dryden C, Wilkinson J, Young D, Brooker RJ; Scottish Paediatric Cystic Fibrosis Managed Clinical Network (SPCFMCN). The impact of 12 months treatment with ivacaftor on Scottish paediatric patients with cystic fibrosis with the G551D mutation: a review. Arch Dis Child. 2018 Jan;103(1):68-70. doi: 10.1136/archdischild-2015-310420. Epub 2016 Jun 10.[Pubmed]

Fig. 31 Carol Dryden ResearchGate
Sttatistically significant improvements in FEV1 and body mass index and a reduction in sweat chloride, all comparable with previously published data were observed. The findings also suggested reduced use of intravenous antibiotics and oral antibiotics. No significant adverse effects were observed but a possible association with cataract formation could not be excluded. This review suggests that, in the short term at least, ivacaftor is effective and safe in paediatric patients ≥6 years of age with G551D.
Dr. Carol Dryden (fig.31) is a consultant Paediatrician in Department of Paediatrics, Wishaw General Hospital, Wishaw, UK.
Edmondson C, Davies JC. Predicting the Future of Cystic Fibrosis Lung Disease: Gene Expression Holds Some of the Answers. Ann Am Thorac Soc. 2018 May;15(5):556-557. doi: 10.1513/AnnalsATS.201802-098ED. [Pubmed] ( an editorial relating to the above article In an editorial relating to the paper of Saavendra et al.)
Fig.32 Claire Edmundson Junior Investigator’s Best Award NACFC 2020
A predictive biomarker of pulmonary exacerbations (APE) would be very useful. They discuss work undertaken in this area by a number of groups. In 2013 some 78 blood based biomarkers had been explored – C-reactive protein being the only one used in clinical practice; even this had limitations. They note that Saavenra and colleagues identify 4 definite gene cluster groups at the start of an APE as described in their paper which it is hoped should provide additional information on outlook.
Dr. Claire Edmondson (fig. 32) is a paediatric registrar at the Royal Brompton currently working for a PhD relating to digital monitoring of CF patients at home. Professor Jane Davies is Senior Lecturer at Imperial College and Hon. Consultant in Paediatric Respiratory Medicine at The Royal Brompton, London. She is a leading UK authority on cystic fibrosis and a senior member of the UK Gene Therapy Consortium
Ehre C, Rushton ZL, Wang B, Hothem LN, Morrison CB, Fontana NC, Markovetz MR, Delion MF, Kato T, Villalon D, Thelin WR, Esther CR Jr, Hill DB, Grubb BR, Livraghi-Butrico A, Donaldson SH, Boucher RC.An Improved Inhaled Mucolytic to Treat Airway Muco-Obstructive Diseases.Am J Respir Crit Care Med. 2018 Sep 13. doi: 10.1164/rccm.201802-0245OC. [Epub ahead of print] [Pubmed]

Fig. 33 Camille Ehre
med.unc.edu
Dithiothreitol (DTT) and a novel mucolytic agent, P3001, were directly compared to N-acetylcysteine (NAC) in vitro and both exhibited superior reducing activities. In vivo, P3001 significantly decreased lung mucus burden in βENaC over-expressing mice whereas NAC did not. In NAC-treated CF patients, deposited NAC was rapidly cleared from the lungs and was ineffective on sputum biophysical properties. In contrast, P3001 acted faster and at lower concentrations than NAC, and was more effective than DNase, in CF sputum ex vivo.
The authors consider these results suggest that reducing the viscoelasticity of airway mucus is an achievable therapeutic goal with P-3001 class mucolytic agents.
Dr. Camille Ehre (fig.33) is Assistant Professor at the Marsico Lung Institute, University of North Carolina School of Medicine.
Eising JB, van der Ent CK, Teske AJ, Vanderschuren MM, Uiterwaal CSPM, Meijboom FJ. Young patients with cystic fibrosis demonstrate subtle alterations of the cardiovascular system. J Cyst Fibros. 2018 Feb 2. pii: S1569-1993(18)30003-1. doi: 10.1016/j.jcf.2017.12.009. [Epub ahead of print] [Pubmed]

Fig. 34 Jacobien Eising emedevents.com
Several studies have shown signs of myocardial dysfunction in adult patients, but little is known about onset and development of these changes over time. In this prospective study, cardiac function in children with cystic fibrosis was compared to that of healthy children. 33 children, aged 3-12years, with cystic fibrosis were recruited from the Wilhelmina Children’s hospital and 33 age-matched healthy children were selected from the WHISTLER study, a population-based cohort study. Measurements of lung function, arterial stiffness, and echocardiography (conventional measures and myocardial deformation imaging) were performed. There were no differences in anthropometrics, lung function (?) and blood pressure between the two groups. The cystic fibrosis children had a higher arterial stiffness compared to the healthy children and a reduced right ventricular systolic function when compared to the healthy children. After adjustment for lung function, global strains of both right and left ventricles were significantly lower in the cystic fibrosis group than in healthy children. Systolic strain rate of basal segment of the left ventricle, the mid segment of the right ventricle and the apical septum were significantly lower in the cystic fibrosis children than in healthy controls.
The authors concluded their study suggests that already at a very young age, children with cystic fibrosis show an increased arterial stiffness and some signs of diminished both right and left ventricular function.
Dr. Jacobien B Eising (Fig. 34) is a paediatric cardiologist in the Netherlands. She published data in 2014 on arterial stiffness and lung function in a group of healthy children (Eur Respir J 2014; 44: 530-532)
Fan Z, Perisse IV, Cotton CU, Regouski M, Meng Q, Domb C, Van Wettere AJ, Wang Z, Harris A, White KL, Polejaeva IA. A sheep model of cystic fibrosis generated by CRISPR/Cas9 disruption of the CFTR gene.JCI Insight. 2018 Oct 4;3(19). pii: 123529. doi: 10.1172/jci.insight.123529. [Epub ahead of print] Free full tex [Pubmed]

Fig. 35 zhiqiang fan
linkedIn.com
CF models have been generated in 4 species (mice, rats, ferrets, and pigs) to enhance our understanding of the CF pathogenesis. Sheep may be a particularly relevant animal to model CF in humans due to the similarities in lung anatomy and development in the two species. Here, the authors describe the generation of a sheep model for CF using CRISPR/Cas9 genome editing and somatic cell nuclear transfer (SCNT) techniques. They generated cells with CFTR gene disruption and used them for production of CFTR-/- and CFTR+/- lambs. The newborn CFTR-/- sheep developed severe disease consistent with CF pathology in humans. Of particular relevance were pancreatic fibrosis, intestinal obstruction, and absence of the vas deferens. Also, substantial liver and gallbladder disease may reflect CF liver disease that is evident in humans. The phenotype of CFTR-/- sheep suggests this large animal model will be a useful resource to advance the development of new CF therapeutics. Moreover, the generation of specific human CF disease-associated mutations in sheep may advance personalized medicine for this common genetic disorder.
Zhiqiang Fan (fig. 35) holds a PostDoc position in the Department of Animal, Dairy and Veterinary Sciences, Utah State University.
Philip Farrell, Claude Férec, Milan Macek, Thomas Frischer, Sabine Renner, Katherina Riss, David barton, Teresa Repetto, maria Tzetis, Karine Giteau, Morton Duno Melissa Rogers, Hara Levy, Mourad Sahbatou, Yann Fichu, Cedreic Le Marechal , Emmanuelle Genin. Estimating the age of p.(Phe508del) with family studies of geographically distinct European populations and the early spread of cystic fibrosis. Eur J Hum Genet 2018 Dec; 26(12):11832-1839. Free PMC article pubmed.ncbi.nlm.nih.gov/30089827/

Fig. 36 Philip Farrell pediatrics.wisc.edu
The high incidence of cystic fibrosis (CF) is due to the frequency of the c.1521_1523delCTT variant in the cystic fibrosis transmembrane conductance regulator (CFTR), but its age and origin are uncertain. This gap limits attempts to shed light on the presumed heterozygote selective advantage that accounts for the variant’s high prevalence among Caucasian Europeans and Europe-derived populations. In addition, explaining the nature of heterozygosity to screened individuals with one c.1521_1523delCTT variant is challenging when families raise questions about these issues. To address this gap, we obtained DNA samples from 190 patients bearing c.1521_1523delCTT and their parents residing in geographically distinct European populations plus a Germany-derived population in the USA. We identified microsatellites spanning CFTR and reconstructed haplotypes at 10 loci to estimate the time/age of the most recent common ancestor (tMRCA) with the Estiageprogram. We found that the age estimates differ between northwestern populations, where the mean tMRCA values vary between 4600 and 4725 years, and the southeastern populations where c.1521_1523delCTT seems to have been introduced only about 1000 years ago. The tMRCA values of Central Europeans were intermediate. Thus, our data resolve a controversy by establishing an early Bronze Age origin of the c.1521_1523delCTT allele and demonstrating its likely spread from northwest to southeast during ancient migrations. Moreover, taking the archeological record into account, our results introduce a novel concept by suggesting that Bell Beaker folk were the probable migrating population responsible for the early dissemination of c.1521_1523delCTT in prehistoric Europe.
2018 Philip Farrell. Discovering the ancient origin of cystic fibrosis, the most common genetic disease in Caucasians. The Conversation. 2018 September 7 (based on above article) https://theconversation.com/discovering-the-ancient-origin-of-cystic-fibrosis-the-most-common-genetic-disease-in-caucasians-100499
Flume PA, Wainwright CE, Elizabeth Tullis D, Rodriguez S, Niknian M, Higgins M, Davies JC, Wagener JS. Recovery of lung function following a pulmonary exacerbation in patients with cystic fibrosis and the G551D-CFTR mutation treated with ivacaftor. J Cyst Fibros. 2018 Jan;17(1):83-88. doi: 10.1016/j.jcf.2017.06.002. Epub 2017 Jun 24.[Pubmed

Fig 37 Patrick Flume providers.muscchealth.org
Pulmonary exacerbations (PEx) are associated with acute loss of lung function that is often not recovered after treatment. We investigated lung function recovery following PEx for ivacaftor- and placebo-treated subjects. Short- and long-term pulmonary function recovery data after PEx were summarized from a placebo-controlled trial in 161 cystic fibrosis patients≥12years old with the G551D-CFTR mutation (NCT00909532). Short-term recovery was measured 2 to 8 weeks after treatment, and long-term recovery was determined at the end-of-study, both compared with baseline measured just prior to the PEx. Fewer patients receiving ivacaftor experienced a PEx than patients receiving placebo (33.7% vs. 56.4%; P=0.004) and had a lower adjusted incidence rate of PEx (0.589 vs. 1.382; P<0.001). The proportion of PEx followed by full short-term recovery of per cent predicted forced expiratory volume in 1s was similar (ivacaftor vs. placebo, 57.1% vs. 53.7), as was the proportion of patients having long-term recovery (46.4% vs. 47.7%).Ivacaftor treatment reduces the frequency of PEx but does not improve on the rate of complete lung function recovery after PEx when compared with placebo.
— The authors observe that “adverse outcome attributed to pulmonary exacerbations occur even when addressing the underlying cause of CF and improving CFTR function in the host”. This is to be expected as most of these patients had past “the point of no return” when rescued CFTR function had been overshadowed by chronic inflammation less dependent on CFTR function. Once again emphasising the great importance of treatment before the stage of chronic inflammation is reached.
Patrick Flume (fig 37) is professor in the Departments of Medicine and Pediatrics, Medical University of South Carolina, 96 Jonathan Lucas St, Room 812-CSB, MSC 630, Charleston, SC 29425, USA.
Foong RE, Harper AJ, Skoric B, King L, Turkovic L, Davis M, Clem CC, Rosenow T, Davis SD, Ranganathan S, Hall GL, Ramsey KA.The clinical utility of lung clearance index in early cystic fibrosis lung disease is not impacted by the number of multiple-breath washout trials. ERJ Open Res. 2018 Feb 16;4(1). pii: 00094-2017. doi: 10.1183/23120541.00094-2017. eCollection 2018 Jan [Pubmed] Free PMC Article

Fig 38 Rachel Foong
ResearchGate
The lung clearance index (LCI) from the multiple-breath washout (MBW) test is a promising surveillance tool for pre-school children with cystic fibrosis (CF). Current guidelines for MBW testing recommend that three acceptable trials are required. However, success rates to achieve these criteria are low in children aged <7 years and feasibility may improve with modified pre-school criteria that accepts tests with two acceptable trials. This study aimed to determine if relationships between LCI and clinical outcomes of CF lung disease differ when only two acceptable MBW trials are assessed. Healthy children and children with CF aged 3-6 years were recruited for MBW testing. Children with CF also underwent bronchoalveolar lavage fluid collection and a chest computed tomography scan. MBW feasibility increased from 46% to 75% when tests with two trials were deemed acceptable compared with tests where three acceptable trials were required. Relationships between MBW outcomes and markers of pulmonary inflammation, infection and structural lung disease were not different between tests with three acceptable trials compared with tests with two acceptable trials. This study indicates that pre-school MBW data from two acceptable trials may provide sufficient information on ventilation distribution if three acceptable trials are not possible
Dr Rachel E Foong (fig.38) is an NHMRC Early Research Career Fellow with the Children’s Health Team, Telethon Kids Institute
Frost F, Dyce P, Nazareth D, Malone V, Walshaw MJ. Continuous glucose monitoring guided insulin therapy is associated with improved clinical outcomes in cystic fibrosis-related diabetes. J Cyst Fibros. 2018 Jun 6. pii: S1569-1993(18)30587-3. doi: 10.1016/j.jcf.2018.05.005. [Epub ahead of print] [Pubmed]

Fig. 39 Freddy Frost
eMedEvents.com
Continuous glucose monitoring (CGM) allows assessment of day to day glycaemic excursions and detects early glucose handling abnormalities that may not be apparent on oral glucose tolerance testing (OGTT). However, there is little published evidence as to whether these early dysglycaemic changes are amenable to treatment. We present outcomes following CGM guided insulin initiation at our centre. Adults without a prior diagnosis of cystic fibrosis related diabetes (CFRD) whom underwent >72 h CGM at our adult CF centre were included in the study. Clinical outcomes including weight and pulmonary function changes over the next 12 months were compared between groups based on CGM results and subsequent management.
CGM profiles for 59 patients were analysed. Insulin was commenced in 37 patients who had evidence of hyperglycaemia on CGM. Significant improvements in mean [95% confidence intervals] forced expiratory volume in 1 s (FEV1) (+4.3% predicted [1.06-7.48], p = 0.01) and weight (+1.2 kg [0.32-2.15], p = 0.01) were observed at 3 months in the insulin group. Annual rate of pulmonary function decline was also improved following insulin initiation. Insulin treatment targeted towards glycaemic excursions seen on CGM is associated with improvements in lung function and weight with subsequent reduced pulmonary function decline.
Freddy Frost (fig. 39) is Liverpool Adult CF Centre, Liverpool Heart and Chest Hospital, Liverpool L14 3PE, UK.
Garcia BA, Carden JL, Goodwin DL, Smith TA, Gaggar A, Leon K, Antony VB, Rowe SM, Solomon GM. Implementation of a successful eradication protocol for Burkholderia Cepacia complex in cystic fibrosis patients. BMC Pulm Med. 2018 Feb 14;18(1):35. doi: 10.1186/s12890-018-0594-8.2 free PMC article[Pubmed]The authors developed and implemented a single center Bcc eradication protocol that included an intensive combination of intravenous, inhaled, and oral antibiotic therapies based on in vitro sensitivities. They conducted a retrospective cohort analysis of clinical outcomes compared to patients with chronic Bcc infection. Six patients were identified as having a newly acquired Bcc colonization and were placed on the eradication protocol. Sequential sputum samples after completion of the protocol demonstrated sustained clearance of Bcc in all patients. Lung function and nutritional status remained stable in the year following eradication.Clearance of Bcc from sputum cultures using a standardized protocol was successful at one year and was associated with clinical stability.
Details of the protocol are included in the free article available via PubMed. Essentially an induction period (21 days) of intravenous tobramycin and ceftazidime, inhaled tobramycin oral trimethoprim/sulphamethoxazole and oral azithromycin then a consolidation period (2 months) of inhaled tobramycin, oral trimethoprim/sulfamethoxazole and azithromycin.
From the Department of Medicine, Division of Allergy, Pulmonary and Critical Care Medicine, 1900 University Blvd, THT 422, Birmingham, AL, 35294, USA.
Garg M, Leach ST, Pang T, Needham B, Coffey MJ, Katz T, Strachan R, Widger J, Field P, Belessis Y, Chuang S, Day AS, Jaffe A, Ooi CY. Age-related levels of fecal M2-pyruvate kinase in children with cystic fibrosis and healthy children 0 to 10years old. J Cyst Fibros. 2018 Jan;17(1):109-113. doi: 10.1016/j.jcf.2017.07.011. Epub 2017 Jul 25. [Pubmed]
The pathogenesis of gut inflammation, bacterial dysbiosis and increased rates of malignancy in CF is unclear. Fecal M2-pyruvate kinase (M2-PK) is a biomarker indicative of cellular proliferation that may be raised in intestinal malignancy and inflammation. Biomarkers, including M2-PK, may be useful in assessing effects of novel therapies on the gastrointestinal tract. M2-PK was measured in stools collected from patients with CF and HC (0-10years). Linear mixed model analysis was used. M2-PK levels did not significantly change in children with CF (36 patients, 77 samples) (P=0.998) or HC (45 patients, 45 samples) (P=0.21), over the age range 0-10years. Patients with CF had elevated M2-PK compared to HC (median [IQR; range]: 10.7 [5.7-28.6; 1.0-239.1] (n=77) vs. 1.0 [1.0-1.0; 1.0-50.0] (n=45) U/mL, respectively; P=0.001).
The authors concluded fecal M2-PK was elevated in children with CF compared with healthy children during infancy and throughout childhood suggesting abnormalities of increased intestinal cellular turnover in the CF gut exist in early life. They also suggest that the test may have a role in assessing the effect of recent therapies.
Millie Garg is at the School of Women and Children’s Health, Medicine, The University of New South Wales, High Street, Sydney Children’s Hospital, Randwick 2031, New South Wales, Australia.
Giannakopoulos A, Katelaris A, Noni M, Karakonstantakis T, Kanaka-Gantenbein C, Doudounakis S.Hyperthyrotropinemia in newly diagnosed cystic fibrosis patients with pancreatic insufficiency reversed by enzyme therapy. Eur J Pediatr. 2018 May;177(5):775-779. doi: 10.1007/s00431-018-3120-3. Epub 2018 Feb 27. [Pubmed]
Patients with cystic fibrosis (CF) commonly present with an elevated TSH concentration, suggesting subclinical hypothyroidism. Its relation to concomitant pancreatic insufficiency and its natural course upon initiation of enzyme replacement have not been adequately studied. Herein, we investigated the thyroid function in newly diagnosed infants with CF and monitored the course of thyroid function response to pancreatic enzyme substitution treatment. Fourteen, newly diagnosed infants with CF and pancreatic insufficiency, were followed every 6-8 weeks for 6 months ensuing onset of pancreatic enzyme substitution therapy. All infants had normal TSH values on neonatal screening. Ten out of 14 (71%) had hyperthyrotropinemia and normal freeT4 values at presentation. No patient received thyroxine. Upon follow-up, after 6 months, TSH values normalized in 90% of infants with CF and hyperthyrotropinemia. Serum selenium levels were negatively correlated with TSH levels.
The authors conclude mild TSH elevation is a frequent finding in newly diagnosed cystic fibrosis patients with pancreatic insufficiency during infancy. TSH elevation resolves in most cases after initiation of enzyme substitution and improvement of nutritional status without any substitutive therapy with thyroxine.
Dr Aris Giannakopoulos, is a paediatric endocrinologist at the University Hospital of Patras, Greece; the other authors are from The Cystic Fibrosis Unit, “Aghia Sophia” Children’s Hospital, Athens
Goldenberg RB. Singing Lessons for Respiratory Health: A Literature Review. J Voice. 2018 Jan;32(1):85-94. doi: 10.1016/j.jvoice.2017.03.021. Epub 2017 Apr 29. Open access to article in J. Voice [Pubmed]

Fig. 40 Rachel Goldenberg
Several studies have explored the role of music and singing as a treatment for respiratory symptoms. The objective of this paper was to review the current body of literature in regard to the use of singing as both a physiological and a psychological therapy for respiratory disease and assess the role the singing teacher might play in this treatment. This is a literature review, discussion of results and directions for further research. Multiple databases were searched using keywords such as “respiratory,” “physiotherapy,” and “pulmonary” in conjunction with “singing.” Studies that met selection criteria Seventeen studies pertaining to multiple conditions including chronic obstructive pulmonary disease, asthma, cystic fibrosis, cancer, Parkinson disease, quadriplegia, and multiple sclerosis were analysed. All studies reported trends of positive physical and/or quality of life outcomes after a series of singing lessons, regardless of statistical significance. Several noted improvements in maximum expiratory pressure and overall breathing technique. Many studies included open-ended interviews revealing participants’ perception of singing as an effective therapy that was fun, improved mood, taught breathing and breath control, was a good exercise for the lungs, and had improved physical functioning
The author concluded that singing can be used as an adjunctive treatment for respiratory disease, with the best results occurring after long-term study. Group lessons and a strong teacher relationship feed the need for social interaction and support, which can facilitate treatment compliance. Further research is warranted.
— This is an interesting extensive review of the previous literature by Rachel Goldenberg of the Department of Music, Ambrose University, Calgary, Canada. The publications concerning CF in both children and adults are reviewed in some detail concluding there appeared to be overall benefit.
Dr. Rachel (Brager) Goldenberg (fig.40) is a distinguished Canadian soprano whose doctoral thesis was focused on the use of singing lessons as an adjunctive airway clearance technique for Cystic Fibrosis. She has presented her dissertation research and continuing research in the use of singing for respiratory ailments across the United States, Canada and Europe.
Gorrie A, Archibald AD, Ioannou L, Curnow L, McClaren B. Exploring approaches to facilitate family communication of genetic risk information after cystic fibrosis population carrier screening. J Community Genet. 2018 Jan;9(1):71-80. doi: 10.1007/s12687-017-0337-1. Epub 2017 Oct 2. Free PMC Article [Pubmed]

Fig. 41 Anita Gorrie
X.com
Population carrier screening for cystic fibrosis (CF) enables individuals with no known family history of the condition to ascertain their risk of having a child with CF. When an individual is identified as a carrier of CF, a life-shortening condition, they are encouraged to inform their relatives who are at increased risk of being a carrier. Research suggests that the uptake of CF carrier testing amongst relatives of carriers or people with CF is low. This study aimed to explore approaches to facilitate the process of family communication of genetic information after an individual is identified as a carrier of CF through population screening. Five key informants were interviewed to inform the development of a telephone survey which was administered to 21 individuals identified as carriers of CF through population carrier screening at Victorian Clinical Genetics Services.
This study suggests that providing carriers with additional information and follow-up support would be appreciated by carriers and could result in more accurate information being disseminated more widely within families, which could lead to more at-risk relatives accessing testing. Suggested strategies to enhance current practice include mailing a fact sheet to carriers and a follow-up telephone call provided by a genetic counsellor to carriers to offer further support in communicating this information to their relatives.
Anita Gorrie (fig.41) is a Genetic Counsellor in the Department of Paediatrics, The University of Melbourne, Melbourne, VIC, Australia.
Graeber SY, Dopfer C, Naehrlich L, Gyulumyan L, Scheuermann H, Hirtz S, Wege S, Mairbäurl H, Dorda M, Hyde R, Bagheri-Hanson A, Rueckes-Nilges C, Fischer S, Mall MA, Tümmler B. Effects of Lumacaftor/Ivacaftor Therapy on CFTR Function in Phe508del Homozygous Patients with Cystic Fibrosis. Am J Respir Crit Care Med. 2018 Jan 12. doi: 10.1164/rccm.201710-1983OC. [Epub ahead of print] [Pubmed]

Fig. 42 Simon Graeber ResearchGate
The phase 3 trials of lumacaftor examined clinical outcomes, but did not evaluate the CFTR function in patients. This study examines the effect of lumacaftor-ivacaftor on biomarkers of CFTR function in Phe508del homozygous treated CF patients aged 12 years and older. A total of 53 patients were enrolled in the study and 52 patients had baseline and follow up measurements. After initiation of lumacaftor-ivacaftor sweat chloride concentrations were reduced by 17.8 mmol/L (IQR -25.9 to -6.1; p<0.001), NPD showed partial rescue of CFTR function in nasal epithelia to a level of 10.2% (IQR 0.0 to 26.1;p<0.011), and ICM showed functional improvement in rectal epithelia to a level of 17.7% of normal (IQR 10.8 to 29.0; p<0.001). All patients improved in at least one CFTR biomarker, but no correlations were found between CFTR biomarker responses and clinical outcomes.
So lumacaftor-ivacaftor results in partial rescue of Phe508del CFTR function to levels comparable to the lower range of CFTR activity found in patients with residual function mutations. Functional improvement was detected even in the absence of short-term improvement of FEV1 % predicted and BMI.
Dr Simon Graeber (fig. 42) is a paediatric pulmonologist at the University of Heidelberg.
Gregg AR. Expanded Carrier Screening. Obstet Gynecol Clin North Am. 2018 Mar;45(1):103-112. doi: 10.1016/j.ogc.2017.10.005. [Pubmed]

Fig. 43 Anthony R Gregg LinkedIn.com
Prenatal carrier screening has expanded to include a larger number of genes and variants offered to all couples considering or with an on going pregnancy. Panethnic screening for cystic fibrosis and spinal muscular atrophy and screening for a limited number of conditions based on ethnicity are recommended by the American College of Obstetricians and Gynecologists. Residual risk calculations have become an obsolete part of post-test counselling when expanded carrier screening (ECS) is selected. The Perception of Uncertainties in Genome Sequencing scale offers a useful understanding of the pre-test and post-test counselling concerns that should be considered as part of ECS implementation.
Dr. Anthony Gregg (fig.43) is in the Department of Obstetrics and Gynecology, University of Florida College of Medicine, Gainesville.
Anthony R Gregg, Janice G Edwards. Prenatal genetic carrier screening in the genomic age. Semin Perinatol. 2018 Aug;42(5):303-306.doi: 10.1053/j.semperi.2018.07.019.Epub 2018 Jul 26. [Pubmed]

Fig. 44 Janice Edwards sc.edu/study
The Society for Maternal-Fetal Medicine, American College of Obstetricians and Gynecologists (ACOG), and American College of Medical Genetics and Genomics (ACMG) held a workshop entitled “Prenatal Genetic Testing” on January 25, 2017 to address several questions arising from the increasing implementation of preconception and prenatal expanded carrier screening (ECS). ECS allows for identification of a greater number of genetic sequencing changes (not all of which cause disease) and simultaneous testing for an increased number of genetic conditions without limitation to specific ethnic groups.
The workshop participants reached consensus on the following: ethnicity based testing cannot be completely abandoned in favor of panethnic ECS; the specific approach to screening should be a patient’s choice and not driven solely by provider preference; organizations should work to develop a framework for vetting conditions that should be reported on ECS panels; compared with prenatal screening, preconception screening is ideal and, at this time, due to the costs and the need for timeliness associated with prenatal screening, post-test counselling and testing, that when ECS is offered it should be presented as a preconception option; preconception and prenatal panels should be identical across the spectrum of patients, including those undergoing assisted reproduction; adult-onset conditions should not be included on ECS panels; partners should be offered next-generation sequencing to identify rare variants when the first partner screened is determined to be a carrier; re-screening in subsequent pregnancies is not indicated, despite the potential for expansion of carrier screening conditions and variants; and more education about ECS for providers and patients is necessary to implement prenatal carrier screening in a responsible way.
Anthony Gregg, (fig. 43) Chief Obstetrics and Gynecology, Maternal Fetal Medicine, Baylor University Medical Center, 3500 Gaston Ave., Dallas, and President of the American College of Medical Genetics and Genomics.
Janice Edwards (fig. 44) Director of the Genetic Counseling Program, School of Medicine, University of South Carolina, Columbia,
Guimbellot JS, Acosta EP, Rowe SM. Sensitivity of ivacaftor to drug-drug interactions with rifampin, a cytochrome P450 3A4 inducer. Pediatr Pulmonol. 2018 May;53(5):E6-E8. doi: 10.1002/ppul.23971. Epub 2018 Feb 28.[Pubmed]
The CFTR potentiator ivacaftor is responsible for significant clinical improvements among a subset of patients with cystic fibrosis. Because it is a substrate of the cytochrome P450 system, specifically CYP3A4/5, ivacaftor is subject to significant drug-drug interactions, including due to commonly used antimicrobials such as rifampin. While the interaction of rifampin and ivacaftor has been examined in vitro, severe adverse events resulting from this interaction have not been reported in the literature. In this report, the authors describe the termination of steady, long-term improvement in a patient taking ivacaftor, resulting from the use of rifampin and precipitating a significant pulmonary exacerbation.
Guimbello J, Solomon GM, Baines A, Heltshe SL, VanDalfsen J, Joseloff E, Sagel SD, Rowe SM; GOALe(2) Investigators. Effectiveness of ivacaftor in cystic fibrosis patients with non-G551D gating mutations J Cyst Fibros 2018 Apr 21. pii: S1569-1993(18)30090-0. doi: 10.1016/j.jcf.2018.04.004. [Epub ahead of print] [Pubmed]

Fig. 45 Jennifer Guimbelllot
childrensal.org
The authors report the results of an observational study investigating its effects in CF patients with non-G551D gating mutations. Patients with non-G551D gating mutations were recruited to an open-label study evaluating ivacaftor. Primary outcomes included: leven Rowe ung function, sweat chloride, weight gain, and quality of life scores.
Twenty-one subjects were enrolled and completed 6 months follow-up on ivacaftor; mean age was 25.6 years with 52% <18. Baseline ppFEV1 was 68% and mean sweat chloride 89.6 mEq/L. Participants experienced significant improvements in ppFEV1 (mean absolute increase of 10.9% 95% CI = [2.6,19.3], p = 0.0134), sweat chloride (-48.6 95% CI = [-67.4,-29.9], p < 0.0001), and weight (5.1 kg, 95% CI = [2.8, 7.3], p = 0.0002).
The authors concluded patients with non-G551D gating mutations experienced improved lung function, nutritional status, and quality of life. This study supports on going use of ivacaftor for patients with these mutations.
Jennifer Guimbellot (fig.45) is Assistant Professor and Associate Scientist at the Gregory Fleming James Cystic Fibrosis Research Center, University of Alabama at Birmingham (UAB), Birmingham, AL, USA; Department of Pediatrics, Division of Pulmonary and Sleep Medicine, UAB, Birmingham, AL, USA.
Hadjiliadis D, Khoruts A, Zauber AG, Hempstead SE, Maisonneuve P, Lowenfels AB; Cystic Fibrosis Colorectal Cancer Screening Task Force. Cystic Fibrosis Colorectal Cancer Screening Consensus Recommendations.Gastroenterology. 2018 Feb;154(3):736-745.e14. doi: 10.1053/j.gastro.2017.12.012. Epub 2017 Dec 29.[Pubmed]

Fig. 46. Denis Hadjiliadis pennmediicn.org
The risk of colorectal cancer (CRC) in adults with CF is 5-10 times greater compared to the general population, and 25-30 times greater in CF patients after an organ transplantation. To address this risk, the CF Foundation convened a multi-stakeholder task force to develop CRC screening recommendations. The task force made 10 CRC screening recommendations. They recommend colonoscopy as the preferred screening method, initiation of screening at age 40 years, 5-year re-screening and 3-year surveillance intervals (unless shorter interval is indicated by individual findings), and a CF-specific intensive bowel preparation. Organ transplant recipients with CF should initiate CRC screening at age 30 years within 2 years of the transplantation because of the additional risk for colon cancer associated with immunosuppression. These recommendations aim to help CF adults, families, primary care physicians, gastroenterologists, and CF and transplantation centres address the issue of CRC screening.
Dr. Denis Hadjiliadis (fig. 46) is a pulmonologist in Philadelphia, Pennsylvania and is affiliated with multiple hospitals in the area, including Hospitals of the University of Pennsylvania-Penn Presbyterian and Penn Presbyterian Medical Center and oversees one of the largest adult CF clinics in N America.
Hammond JA, Connett GJ.The use of lumacaftor/ivacaftor to treat acute deterioration in paediatric cystic fibrosis. Paediatr Respir Rev. 2018 Jun;27:16-17. doi: 10.1016/j.prrv.2018.05.008. Epub 2018 May 19. [Pubmed] 29914743

Fig. 47 Gary Connett Child Health International
The authors describe the use of lumacaftor/ivacaftor to treat an 8 year old girl with an unusually severe exacerbation of CF lung disease and review the potential of lumacaftor/ivacaftor as a rescue therapy in the paediatric CF population.
The patient was homozygous for the Phe508del genotype. Her background included cleft palate repair for Pierre Robin sequence, gastrostomy for poor nutrition and fundoplication for severe reflux. She had previously received three courses of intravenous (IV) antibiotics for increased respiratory symptoms, but remained free of chronic infection with Pseudomonas aeruginosa. She was on regular rhDNase and enjoyed normal levels of physical activity. Before her acute severe illness FEV1s were >80%predicted.
She presented with a two week history of deteriorating chest infection FEV1 44%. Despite intensive investigation and treatment at 4 months with an FEV1 of 31% she was referred for transplant assessment and lumacaftor/ivacaftor started.
Within two weeks of commencement there were significant clinical improvements. Oxygen saturations became normal in air and she was discharged within two weeks after discontinuing IV antibiotics. No other changes were made to her treatment. Her FEV1 continued to improve to 80% predicted 1 month after starting treatment and has been maintained at this level for a further 6 months.
Prof. Gary Connett (fig.47) the corresponding author, was appointed as a paediatric respiratory consultant in Southampton in 1995. He is visiting professor to University Hospitals of Latvia (2009) and Singapore (2011) and has held a personal chair as professor of paediatrics at the University of Southampton since 2018.
— This is remarkable story. Professor Connett kindly informed me (in December 2018) that the child has made good progress maintaining an FEV1 between 60-70% predicted and having three monthly courses of intravenous and antibiotics. She has a relatively good quality of life attending school and participating in sporting activities. She does however feel precarious and at risk of further deterioration
Harper JC,. Preimplantation genetic screening. J Med Screen 12018 Mar; 24(1):1-5. [Pubmed]

Fig. 48 Joyce Harper UCL Profiles
Preimplantation genetic diagnosis was first successfully performed in 1989 as an alternative to prenatal diagnosis for couples at risk of transmitting a genetic or chromosomal abnormality, such as cystic fibrosis, to their child. From embryos generated in vitro, biopsied cells are genetically tested. From the mid-1990s, this technology has been employed as an embryo selection tool for patients undergoing in vitro fertilisation, screening as many chromosomes as possible, in the hope that selecting chromosomally normal embryos will lead to higher implantation and decreased miscarriage rates. This procedure, preimplantation genetic screening, was initially performed using fluorescent in situ hybridisation, but 11 randomised controlled trials of screening using this technique showed no improvement in in vitro fertilisation delivery rates. Progress in genetic testing has led to the introduction of array comparative genomic hybridisation, quantitative polymerase chain reaction, and next generation sequencing for preimplantation genetic screening, and three small randomised controlled trials of preimplantation genetic screening using these new techniques indicate a modest benefit. Other trials are still in progress but, regardless of their results, preimplantation genetic screening is now being offered globally. In the near future, it is likely that sequencing will be used to screen the full genetic code of the embryoA study
Joyce Harper (fig.48) is Professor of Human Genetics and Embryology at the Institute for Women’s Health, University College London, London, UK.
Harun SN, Wainwright CE, Grimwood K, Hennig S; Australasian Cystic Fibrosis Bronchoalveolar Lavage (ACFBAL) study group. Aspergillus and progression of lung disease in children with cystic fibrosis.Thorax. 2018 Oct 1. pii: thoraxjnl-2018-211550. doi: 10.1136/thoraxjnl-2018-211550. [Epub ahead of print] [Pubmed]

Fig. 49 Sabariah Noor Harun
pha.usm.my
A study to determine if positive respiratory cultures of Aspergillus species are associated with: (1) increased structural lung injury at age 5 years; (2) accelerated lung function decline between ages 5 years and 14 years and (3) to identify explanatory variables. Positive Aspergillus BAL cultures at age 5 years were significantly associated with increased HRCT scores for air trapping (OR 5.53, 95% CI 2.35 to 10.82). However, positive Aspergillus cultures were not associated with either FEV1% predicted at age 5 years or FEV1% predicted by age following adjustment for body mass index z-score and hospitalisation secondary to pulmonary exacerbations. Lung function demonstrated a non-linear decline in this population.
The authors concluded that in children with cystic fibrosis, positive Aspergillus BAL cultures at age 5 years were associated contemporaneously with air trapping but not bronchiectasis. No association was observed between positive Aspergillus BAL cultures on FEV1% predicted at age 5 years or with lung function decline between ages 5 years and 14 years.
Dr Sabariah Noor Harun (fig.49) is a lecturer at the School of Pharmaceutical Sciences, Univsiti Sains Malaysia, Penang and the School of Pharmacy, University of Queensland
Héry-Arnaud G, Boutin S, Cuthbertson L, Elborn SJ, Tunney MM.The lung and gut microbiome: what has to be taken into consideration for cystic fibrosis? J Cyst Fibros. 2018 Nov 25. pii: S1569-1993(18)30931-7. doi: 10.1016/j.jcf.2018.11.003. [Epub ahead of print] [Pubmed]
-

Fig. 50 Genevieve Hery-Arnaud ResearchGate
The 15th European Cystic Fibrosis Society (ECFS) Basic Science pre-conference Symposium focused on the topic of the microbiome, asking the question “The lung and gut microbiome: what has to be considered for cystic fibrosis (CF)?” This review gives an overview of the main points raised during the symposium, which dealt with the technical considerations, pathophysiology and clinical implications of the microbiome in CF.
Dr Genevieve Hery-Arnaud (fig.50) is a scientist at Université de Bretagne Occidentale / Centre Hospitalier Régional Universitaire de Brest, France
Ho LP, Samways JM, Porteous DJ, Dorin JR, Carothers A, Greening AP, Innes JA. Correlation between nasal potential difference measurements, genotype and clinical condition in patients with cystic fibrosis. Eur Respir J. 1997 Sep;10(9):2018-22. Free article. [Pubmed]
- In cystic fibrosis (CF), the clinical condition of patients correlates poorly with genotype. One possible explanation is that clinical status is influenced by net preserved chloride secretion rather than the CF mutation. The authors tested the relationships between residual chloride secretion, as measured by nasal potential difference (PD) and the type of mutation (genotypes expressing apical cystic fibrosis transmembrane conductance regulator (CFTR) protein versus those that do not) and clinical status. Twenty-two CF patients (mean age 25.7 yrs, 11 females and 11 males, mean forced expiratory volume in one second (FEV1) 53.1% of predicted) with defined genotypes were recruited. Nasal PD was measured using a standard protocol involving the perfusion of the nasal epithelium with a sodium channel blocker (amiloride), followed by a solution of low chloride and finally with isoprenaline.
Patients with apical CFTR protein showed higher residual chloride secretion than those without (amiloride to isoprenaline value of 4.59 and 0.56 mV, respectively, p = 0.01). There was no correlation between mutation type and clinical condition. When these patients were re-categorized as “high” (> 10 mV amiloride to isoprenaline response) or “low” (10 mV or less) chloride secretors, the authors found that the former group had a significantly higher FEV1 (67.7 versus 48.3% pred) and a better pulmonary radiological score (4.14 versus 7.07, by Northern scoring system). These results suggest that some cystic fibrosis patients, regardless of genotype, have an ability to secrete chloride when stimulated with chloride secretatagogues, and this is correlated with a better lung function. These results also have implications for the use of potential difference measurements in novel cystic fibrosis transmembrane conductance regulator replacement trials.
-

- Fig.53 Julia Dorin
-

- Fig.52 Alistair Innes
-

- Fig. 51 David Porteous
-

- Fig. 54 Andrew Greening
— This study comes from the clinicians and scientists of the major adult CF service in Scotland (services.nhslothian.scot). The service was developed in 1992 by Professor Andy Greening (fig.54) and the late Dr. Sandy Raeburn with a Clinical CF Fellow and a CF Liaison Sister and the support of the CF Trust. They convinced the Scottish Office of the need for Specialist CF Centres and were permitted to set up the Scottish Adult CF Service. By 2017 the clinic is led by Dr. Alistair Innes (fig.52), the original number of 55 patients had increased to 205 plus another 48 outreach patients in Dundee. Some attend clinics in Edinburgh, others attend clinics in Inverness, Dumfries and Dundee.
The senior scientists in this publication have also made major contributions to the understanding and progress of cystic fibrosis care. Dr. Julia Dorin (fig.53) created one of the first mice with CF and Professor David Porteous (fig.51) has been a leading member of the UK Gene Therapy Consortium since its formation in 2000.
Hoch H, Sontag MK, Scarbro S, Juarez-Colunga E, McLean C, Kempe A, Sagel SD. Clinical outcomes in U.S. infants with cystic fibrosis from 2001 to 2012. Pediatr Pulmonol. 2018 Sep 26. doi: 10.1002/ppul.24165. [Epub ahead of print] pubmed.ncbi.nlm.nih.gov/30259702/

Fig 55 Heather Hoch
Doximetry
All 50 United States implemented newborn screening (NBS) for cystic fibrosis (CF) by 2010. The purpose of this study was to evaluate trends over the decade when NBS became universal to determine current rates of malnutrition, stunting, and infection rates in U.S. infants with CF.Data were obtained on 8178 infants diagnosed with CF. The percentage of infants diagnosed by NBS increased from 15% in 2001-83% in 2012 (P < 0.001).
The authors concluded nationwide implementation of CF NBS is temporally associated with significant improvements in growth outcomes and reductions in P. aeruginosa infections. However, current rates of malnutrition, stunting, and airway infection present a target for early intervention and quality improvement efforts.
Heather Hoch (Fig.55) is at the Department of Pediatrics, University of Colorado School of Medicine and Children’s Hospital Colorado, Aurora, Colorado.
Holtkamp KCA, Henneman L, Gille JJP, Meijers-Heijboer H, Cornel MCLakeman P. Direct-to-consumer carrier screening for cystic fibrosis via a hospital website: a 6-year evaluation. J Community Genet. 2018 Sep 18. doi: 10.1007/s12687-018-0388-y. [Epub ahead of print] [Pubmed]

Fig. 56 Kim Holtkamp
ResearchGate
A Dutch university hospital started offering cystic fibrosis (CF) carrier screening directly to consumers (DTC) through their website in 2010. A 6-year process evaluation was conducted to evaluate the offer. Screening was implemented as intended. However, uptake was lower than expected. Forty-four tests have been requested, partly by couples with a positive family history for CF, which was not the intended target group. Users were generally positive about the screening offer, citing accessibility, ease of testing, anonymity, and perceived shortcomings of regular healthcare as reasons for requesting screening.
DTC CF carrier screening via a university hospital website is feasible, but is seldom used.
Considering technological advances, continuation of this specific offer is questionable.
Dr Kim Holtkamp (fig.56) is a Researcher in Community Genetics Department of Clinical Genetics, VU University Medical Center, PO Box 7057, 1007 MB, Amsterdam, The Netherlands.
Hoo ZH, Edenborough FP, Curley R, Prtak L, Dewar J, Allenby MI6, Nightingale JA, Wildman MJ. Understanding Pseudomonas status among adults with cystic fibrosis: a real-world comparison of the Leeds criteria against clinicians’ decision.Eur J Clin Microbiol Infect Dis. 2018 Jan 6. doi: 10.1007/s10096-017-3168-4. [Epub ahead of print] [Pubmed]

Fig. 57 Zhe Hui Hoo
ResearchGate
Pseudomonas aeruginosa status influences cystic fibrosis (CF) clinical management but no ‘gold standard’ definition exists. The Leeds criteria are commonly used but may lack sensitivity for chronic P. aeruginosa. We compared clinicians’ decision with the Leeds criteria in three adult CF centres. Two independent prospective datasets (Sheffield dataset, n = 185 adults; ACtiF pilot dataset, n = 62 adults from two different centres) were analysed. Clinicians involved in deciding P. aeruginosa status were blinded to the study objectives. Clinicians considered more adults with CF to have chronic P. aeruginosa infection compared to the Leeds criteria. This was more so for the Sheffield dataset (106/185, 57.3% with clinicians’ decision vs. 80/185, 43.2% with the Leeds criteria; kappa coefficient between these two methods 0.72) compared to the ACtiF pilot dataset (34/62, 54.8% with clinicians’ decision vs. 30/62, 48.4% with the Leeds criteria; kappa coefficient between these two methods 0.82). However, clinicians across different centres were relatively consistent once age and severity of lung disease, as indicated by the type of respiratory samples provided, were taken into account. Agreement in P. aeruginosa status was similar for both datasets among adults who predominantly provided sputum samples (kappa coefficient 0.78) or adults > 25 years old (kappa coefficient 0.82). Across three different centres, clinicians did not always agree with the Leeds criteria and tended to consider the Leeds criteria to lack sensitivity. Where disagreement occurred, clinicians tended to diagnose chronic P. aeruginosa infection because other relevant information was considered. These results suggest that a better definition for chronic P. aeruginosa might be developed by using consensus methods to move beyond a definition wholly dependent on standard microbiological results.
— It is surprising that the authors make virtually no mention of Pseudomonas antibodies – shown in numerous previous studies to be an accurate indication of chronic Pseudomonas infection since the early classic studies of Niels Hoiby in the Seventies (Hoiby N et al, Acta Path Microbiol Scand 1973; 81:298-308. [PubMed]; Hoiby N, Acta Pathol Microbiol Immunol Scand 1977; Supplement (262): 1-96. [PubMed])and indeed shown to correlate with the presence of chronic Pseudomonas infection in the original paper describing the Leeds criteria (Lee TWR et al.J Cyst Fibros 2003; 2:29-34.[PubMed].
Dr Zhe Hui Hoo (fig.57) is Clinical Research Fellow, in the Medical Statistics Group, University of Sheffield.
Hubert D, Dehillotte C, Munck A, David V, Baek J, Mely L, Dominique S, Ramel S, Danner Boucher I, Lefeuvre S, Reynaud Q, Colomb-Jung V, Bakouboula P, Lemonnier L.Retrospective observational study of French patients with cystic fibrosis and a Gly551Asp-CFTR mutation after 1 and 2years of treatment with ivacaftor in a real-world setting. J Cyst Fibros. 2018 Jan;17(1):89-95. doi: 10.1016/j.jcf.2017.07.001. Epub 2017 Jul 12. [PubMed].
-

Fig. 58 Dominique Hubert écorné-cf
A retrospective observational multicentre study collected clinical data in the year before and the 2years after ivacaftor initiation in patients with CF and a Gly551Asp-CFTR mutation. Fifty-seven patients were included. Mean absolute change in FEV1% predicted improved from baseline to Year 1 (8.4%; p<0.001) and Year 2 (7.2%; p=0.006). Statistically significant benefits were observed with increased body mass index, fewer Pseudomonas aeruginosa (56.2-41.3%) and Staphylococcus aureus (75%-58.7%) positive cultures, and decreased IV antibiotics (12.7-5.4 days) and maintenance treatment prescriptions (including azithromycin (47.4-31.3%), Dornase alpha (56.1-39.6%) and nutritional supplements ( 28.1-14.6%). No significant adverse events were reported. The clinical benefits of ivacaftor reported in previous clinical trials were confirmed in a real-world setting two years post-initiation, also reducing treatment burden.
– Useful information with “real world” results. The use of pancreatic enzymes remained unchanged (89.5-87.5%) as one would expect. The unusually high proportion of patients initially infected by S. aureus (75%) and the many receiving inhaled corticosteroids (63.2%) is interesting. The authors speculate as to whether use of ivacaftor could lead to a reduction in other long-term therapies.
Dominique Hubert (fig.58) is a paediatrician in the Pulmonary Department, Adult CF Centre, Cochin Hospital, AP-HP, Paris, France; Université Paris Descartes, Sorbonne Paris Cité, Paris, France.
Hurley MN, Fogarty A, McKeever TM, Goss CH, Rosenfeld M, Smyth AR. Early Respiratory Bacterial Detection and Antistaphylococcal Antibiotic Prophylaxis in Young Children with Cystic Fibrosis. Ann Am Thorac Soc. 2018 Jan;15(1):42-48. doi: 10.1513/AnnalsATS.201705-376OC [PubMed]

Fig.59 Matthew Hurley
ResearchGate
Consensus is lacking regarding anti-staphylococcal antibiotic prophylaxis use for young children with cystic fibrosis. Prophylaxis is recommended in the United Kingdom, but it is recommended against in the United States. To test the hypothesis that anti-staphylococcal antibiotic prophylaxis is associated with a decreased risk of Staphylococcus aureus acquisition but no increased risk of Pseudomonas aeruginosa acquisition. The authors undertook a longitudinal observational study of children with cystic fibrosis who were recruited from birth (or from their first registry entry in the period) and followed until the age of 4 years (1,500 d) using 2000-2009 data from the UK Cystic Fibrosis Trust and Cystic Fibrosis Foundation registries. Children were excluded if they had a positive culture result for S. aureus or P. aeruginosa, or if they were receiving inhaled antibiotics, at the first encounter. Time to first S. aureus and P. aeruginosa detection in the UK/U.S. cohorts was compared using a Cox proportional hazards model. A UK-based analysis compared the same for those receiving flucloxacillin with those who received no prophylaxis. They included the following covariates: sex, age at registry entry, dornase alfa use, genotype, and centre size.
The primary analysis comprised 1,074 UK and 3,677 U.S. children. The risk of first detection was greater in U.S. children than in UK children for S. aureus (hazard ratio [HR], 5.79; 95% confidence interval [CI], 4.85, 6.90; P < 0.001) and P. aeruginosa (HR, 1.92; 95% CI, 1.65, 2.24; P < 0.001). In the UK analysis, we compared 278 children receiving flucloxacillin and 306 receiving no prophylaxis. Flucloxacillin was not associated with a reduced risk of S. aureus detection (HR, 1.22; 95% CI, 0.74, 2.0; P = 0.43), but it was associated with an increased risk of P. aeruginosa detection (HR, 2.53; 95% CI, 1.71, 3.74; P < 0.001). None of the covariates significantly affected the risk estimate in either analysis.
The risk of first detection of S. aureus and P. aeruginosa was greater in the United States than in the United Kingdom. In the United Kingdom, the risk of first P. aeruginosa detection was increased among those receiving flucloxacillin compared with those who received no prophylaxis. The authors consider these observational findings should be examined in randomized controlled trials.
Matthew Hurley (fig.59) is in the Division of Child Health, Obstetrics & Gynaecology, University of Nottingham, and Nottingham Children’s Hospital, Nottingham, United Kingdom.
Hussain N, Hussain F, Malik A, Rizvi M, Patel P, Chittivelu S. A devastating cardiovascular event in an adult cystic fibrosis patient: An unforeseen outcome of increasing life expectancy. Respir Med Case Rep. 2018 Sep 21;25:233-234. doi: 10.1016/j.rmcr.2018.09.013. eCollection 2018. Free full text [Pubmed]
-

Fig. 60 Noreen Hussain
As the life expectancy of CF patients has increased, it is important to consider other co-morbidities that these patients may encounter, and the impact this may have on their morbidity and mortality. We present a case of a 33-year-old male admitted to the hospital for a CF exacerbation who had an acute neurological decompensation due to an infarction of his right occipital and posterior temporal lobe.
Noreen Hussain (fig. 60) is at the Department of Internal Medicine; University of Illinois College of Medicine at Peoria, USA.
Jain K, Wainwright C, Smyth AR. Bronchoscopy-guided antimicrobial therapy for cystic fibrosis. Cochrane Database Syst Rev. 2018 Sep 17;9:CD009530. doi: 10.1002/14651858.CD009530.pub4. [Pubmed]

Fig. 61 Kamini Jain
LinkedIn
(Update of Bronchoscopy-guided antimicrobial therapy for cystic fibrosis. [Cochrane Database Syst Rev. 2016]) The authors’ summary states this review, limited to a single, well-designed randomized controlled study (that of Claire Wainwright and colleagues), which shows no clear evidence to support the routine use of bronchoalveolar lavage for the diagnosis and management of pulmonary infection in pre-school children with cystic fibrosis compared to the standard practice of providing treatment based on results of oropharyngeal culture and clinical symptoms. No evidence was available for adult and adolescent populations.
Kamini Jain (fig.61) is in the Division of Child Health, School of Clinical Sciences, University of Nottingham, E Floor, East Block, Queen’s Medical Centre, Derby Road, Nottingham, UK, NG9 2SJ. Subsequently Paediatric Respiratory Consultant, Leicester University Hospitals and NHS.
Jardel S, Reynaud Q, Durieu I. Long term extra pulmonary comorbidities after lung transplantation in cystic fibrosis: update of specificities. Clin Transplant. 2018 Apr 26:e13269. doi: 10.1111/ctr.13269. [Epub ahead of print] pubmed.ncbi.nlm.nih.gov/29700855/

Fig. 62 Sabine Jardel ResearchGate
The purpose of the present paper is to provide an update of the non-respiratory comorbidities following lung transplant (LT) in adult patients with CF and their specificities regarding their multi-systemic underlying condition despite their younger age compared to other patients undergoing LT. Diabetes, renal insufficiency, metabolic bone disease, hypertension, liver disease and cancer are the comorbidities considered in this review. The increase of CF adults living with a lung transplant justifies an update of knowledge for this specific situation (prevalence of these complications, underlying risk factors), in order to provide better medical care and establish early diagnosis strategies.
— This is a excellent liberally referenced (87 references) review of the various co-morbidities that can affect people with CF who have lung transplantation. As a result of their review the authors suggest special attention to perioperative glycaemic control regarding the use of corticosteroids and immunosuppressive drugs: a strict glycaemic control regimen after transplantation as well as maintaining the same screening strategy as for all non-diabetic CF patients. With regard to renal function a GFR using a gold standard method is recommended before transplant as well as measures to prevent peri-operative hypo perfusion and a strict monitoring of postoperative calcineurin blood levels. Exercise, vitamin D levels, calcium supplementation and bisphosphonates have been shown to be protective in preventing bone loss. Colorectal cancer and cervical dysplasia may warrant a specific screening strategy even before transplantation in CF patents, which should start earlier.
Sabine Jardel (fig. 62) is in the Department of Internal Medicine, Adult Cystic Fibrosis Care Center, Hospices Civils de Lyon, Lyon, France.
Keating D, Marigowda G, Burr L, Daines C, Mall MA, McKone EF, Ramsey BW, Rowe SM, Sass LA, Tullis E, McKee CM, Moskowitz SM, Robertson S, Savage J, Simard C, Van Goor F, Waltz D, Xuan F, Young T, Taylor-Cousar JL; VX16-445-001 Study Group. VX-445-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles N Engl J Med. 2018 Oct 25;379(17):1612-1620. doi: 10.1056/NEJMoa1807120. Epub 2018 Oct 18. [Pubmed]
-

Fig. 63 Dominic Keating Monash University
VX-445 is a next-generation cystic fibrosis transmembrane conductance regulator (CFTR) corrector designed to restore Phe508del CFTR protein function in patients with cystic fibrosis when administered with tezacaftor and ivacaftor (VX-445-tezacaftor-ivacaftor). The authors evaluated the effects of VX-445-tezacaftor-ivacaftor on Phe508del CFTR protein processing, trafficking, and chloride transport in human bronchial epithelial cells. On the basis of in vitro activity, a randomized, placebo-controlled, double-blind, dose-ranging, phase 2 trial was conducted to evaluate oral VX-445-tezacaftor-ivacaftor in patients heterozygous for the Phe508del CFTR mutation and a minimal-function mutation (Phe508del-MF) and in patients homozygous for the Phe508del CFTR mutation (Phe508del-Phe508del) after tezacaftor-ivacaftor run-in. Primary end points were safety and absolute change in percentage of predicted forced expiratory volume in 1 second (FEV1) from baseline. Results. In vitro, VX-445-tezacaftor-ivacaftor significantly improved Phe508del CFTR protein processing, trafficking, and chloride transport to a greater extent than any two of these agents in dual combination. In patients with cystic fibrosis, VX-445-tezacaftor-ivacaftor had an acceptable safety and side-effect profile. Most adverse events were mild or moderate. The treatment also resulted in an increased percentage of predicted FEV1 of up to 13.8 points in the Phe508del-MF group (P<0.001). In patients in the Phe508del-Phe508del group, who were already receiving tezacaftor-ivacaftor, the addition of VX-445 resulted in an 11.0-point increase in the percentage of predicted FEV1 (P<0.001). In both groups, there was a decrease in sweat chloride concentrations and improvement in the respiratory domain score on the Cystic Fibrosis Questionnaire-Revised.
The authors concluded the use of VX-445-tezacaftor-ivacaftor to target Phe508del CFTR protein resulted in increased CFTR function in vitro and translated to improvements in patients with cystic fibrosis with one or two Phe508del alleles. This approach has the potential to treat the underlying cause of cystic fibrosis in approximately 90% of patients. (Clinical trials NCT03227471 ; and EudraCT number, 2017-000797-11)
Dominic T Keating (fig.63) is Respiratory Physician at the Alfred Hospital · Department of Allergy, Immunology & Respiratory Medicine (AIRmed) Australia, Melbourne.
Andrea Kelly , Diva D De Leon , Saba Sheikh , Devaney Camburn , Christina Kubrak , Amy J Peleckis , Darko Stefanovski , Denis Hadjiliadis , Michael R Rickels , and Ronald C Rubenstein Islet Hormone and Incretin Secretion in Cystic Fibrosis Following 4-months of Ivacaftor Therapy. Am J Respir Cri Care 2018 Aug 21. https://doi-org.rsm.idm.oclc.org/10.1164/rccm.201806-1018OC [Pubmed]
-

Fig. 64 Andrea Kelly chop.edu
- Diabetes is associated with worse cystic fibrosis (CF) outcomes. The CFTR potentiator ivacaftor is suggested to improve glucose homeostasis in individuals with CF. Objective: The authors tested the hypothesis that clinically-indicated ivacaftor would be associated with improvements in glucose tolerance and insulin and incretin secretion. Methods: Oral glucose tolerance tests, mixed meal tolerance tests, and glucose-potentiated arginine tests were compared pre- and 16-weeks post-ivacaftor initiation in CF participants with at least one CFTR gating or conductance mutation. Meal-related 30- (early-phase) and 180-min incremental area under the curves were calculated as responses for glucose, insulin, C-peptide, and incretin hormones, glucagon-like peptide-1 and glucose-dependent insulinotropic polypeptide. First-phase insulin secretion, glucose-potentiation of arginine-induced insulin secretion, and disposition index were characterized by glucose-potentiated arginine tests.
- Results: Twelve subjects (6M/6F; 7 normal/5 abnormal glucose tolerance [AGT], oral glucose tolerance test one-hour glucose>155 and two-hour glucose<200 mg/dL) of median [min-max] age: 13.8y [6.0-42.0], BMI-Z: 0.66 [-2.4-1.9], and FEV1%-predicted: 102 [39-122] completed the study. Glucose tolerance normalized in one AGT subject. Ivacaftor treatment did not alter meal responses except for an increase in early-phase C-peptide (P=0.04). First-phase (P=0.001) and glucose-potentiation of arginine-induced (P=0.027) insulin secretion assessed by acute C-peptide responses improved after ivacaftor treatment. Consistent with an effect on β-cell function, the disposition index relating the amount of insulin secreted for insulin sensitivity also improved (P=0.04)
-
Andrea Kelly (fig.64) is a paediatrician in the Division of Endocrinology and Diabetes and Division of Pulmonary Medicine and Cystic Fibrosis Center, Department of Pediatrics, Children’s Hospital of Philadelphia, Philadelphia, Pennsylvania.
Ruth H Keogh, Rhonda Szczesniak, David Taylor-Robinson, Diana Bilton Up-to-date and projected estimates of survival for people with cystic fibrosis using baseline characteristics: A longitudinal study using UK patient registry data.J Cyst Fibros. 2018 Mar;17(2):218-227.doi: 10.1016/j.jcf.2017.11.019. Epub 2018 Jan 6. Free PMC article [Pubmed]

Fig. 65 Ruth Keogh Twitter.com
Flexible parametric survival modelling of UK CF Registry data from 2011 to 2015, capturing 602 deaths in 10,428 individuals. Survival curves were estimated from birth; conditional on reaching older ages; and projected under different assumptions concerning future mortality trends, using baseline characteristics of sex, CFTR genotype (zero, one, two copies of F508del) and age at diagnosis.
Findings: Male sex was associated with better survival, as was older age at diagnosis, but only in F508del non-homozygotes. Survival did not differ by genotype among individuals diagnosed at birth. Median survival ages at birth in F508del homozygotes were 46years (males) and 41years (females), and similar in non-homozygotes diagnosed at birth. F508del heterozygotes diagnosed aged 5 had median survival ages of 57 (males) and 51 (females). Conditional on survival to 30, median survival age rises to 52 (males) and 49 (females) in homozygotes. Mortality rates decreased annually by 2% during 2006-2015. Future improvements at this rate suggest median survival ages for F508del homozygous babies of 65 (males) and 56 (females).
Interpretation: Over half of babies born today, and of individuals aged 30 and above today, can expect to survive into at least their fifth decade.
Research in context: Previous studies on survival are reviewed and figures are presented.Dr Ruth Keogh (fig.65) is Professor of Biostatistics and Epidemiology at the London School of Hygiene and Tropical Medicine.
— It is good to see the data from the CF Trust analysed so clearly.
Kim J, Davies Z, Dunn C, Wine JJ, Milla C. Ivacaftor restores CFTR-dependent sweat gland fluid secretion in cystic fibrosis subjects with S945L alleles. J Cyst Fibros. 2018 Mar;17(2):179-185. doi: 10.1016/j.jcf.2017.12.005. Epub 2017 Dec 24. 29279204 ??[Pubmed]
-

Fig. 66 Carlos Milla Stanford Medicine
- A study to determine in vivo effects of CFTR modulators on mutation S945L. The authors measured effects of CFTR modulators on CFTR-dependent sweating (‘C-sweat’) in two pancreatic sufficient cystic fibrosis (CF) subjects. S1 (S945L/G542X) took ivacaftor and S2 (S945L/F508del) took ivacaftor+tezacaftor. Sweating was stimulated pharmacologically to produce sequentially both CFTR-independent (methacholine stimulated) M-sweat and C-sweat; and the ratio of these was compared. Sweat secretion was measured with two methods: real time secretory rate quantitative recording and by optically measuring the growth of sweat bubbles under oil from multiple identified glands.
Using the quantitative recorder, they saw zero C-sweat secretion off-drug, but when on-drug the C-sweat responses for both subjects were comparable to those seen in carriers. The on-drug response was further quantified using the sweat bubble method. Each subject again showed robust C-sweat responses, with C-sweat/M-sweat ratios~half of the ratio determined for a cohort of 40 controls tested under identical conditions.
These in vivo results, consistent with prior in vitro findings, indicate that the drug treatments restore near-normal function to S945L-CFTR, and support the use of ivacaftor as a treatment for CF patients who carry this allele.
Corresponding author is Professor Carlos Milla (fig.66) at the Lucille Salter Packard Children’s Hospital, Stanford University.
Kim M, Cai Q, Oh Y.Therapeutic potential of alpha-1 antitrypsin in human disease. Ann Pediatr Endocrinol Metab. 2018 Sep;23(3):131-135. doi: 10.6065/apem.2018.23.3.131. Epub 2018 Sep 28.[Pubmed] Free PMC Article
-

Fig.67 Youngman Oh
Alpha-1 antitrypsin (AAT), an alpha globulin glycoprotein, is a member of the serine protease inhibitor (serpin) superfamily. The clinical significance of AAT is highlighted by AAT deficiency. Genetic deficiency of AAT can present as several neutrophilic diseases associated with emphysema, liver cirrhosis, panniculitis, and systemic vasculitis. Recently, animal and human studies have shown that AAT can control inflammatory, immunological, and tissue-protective responses. In addition, AAT treatment can prevent overt hyperglycemia, increase insulin secretion, and reduce cytokine-mediated apoptosis of pancreatic β-cells in diabetes. These multifunctional roles of AAT draw attention to the glycoprotein’s therapeutic potential for many inflammatory and autoimmune diseases beyond AAT deficiency. As underlying mechanisms, recent studies have suggested the importance of serine protease inhibitory activity of AAT in obesity-associated insulin resistance, chronic obstructive pulmonary disease, and cystic fibrosis. In this review, we explore the multiple functions of AAT, in particular, the anti-inflammatory and serine protease inhibitory functions, and AAT’s therapeutic potential in a variety of human diseases through published literature.
— With regard to alpha-1 antitrypsin for cystic fibrosis, the authors of this article comment as follows- Thus, in CF, the treatment focus is on decreasing neutrophil hyper activation and counteracting the effects of NE on the lung. To inhibit NE in the lung during CF progression, early studies have focused on the augmentation of systemic AAT levels by intravenous injection (McElvaney NG[PubMed]).Recently, a randomized, double-blind, placebo-controlled phase 2a study in CF patients has been further performed to evaluate the safety of 100 or 200 mg of inhaled AAT once daily for 3 weeks in 30 adult subjects and reported that inhalation is safe and well tolerated (Gaggar A et al, 2016 [PubMed]).Further multiple studies have demonstrated that AAT administered by inhalation can control neutrophil function and NE levels in a dose-dependent fashion as well as inflammation in the lung . However, many obstacles still remain in applying AAT in CF since mixed results have been observed depending on the devices used as well as lung condition and NE concentration of patients
Dr. Min Sun Kim is Associate Professor in the Division of Endocrinology, Department of Pediatrics, Chonbuk National University Medical School, Jeonju, Korea.
Corresponding author is Professor Youngman OH,(fig.67) Department of Pathology, School of Medicine Medical College of Virginia Campus, Virginia Commonwealth University, Richmond, VA
Kirwan L, Fletcher G, Harrington M, Jeleniewska P, Zhou S, Casserly B, Gallagher CG, Greally P, Gunaratnam C, Herzig M, Linnane B, McElvaney NG. McKone EF, McNally P, Mullane D, Ní Chróinín M, O’Mahony M, Plant BJ, Jackson AD.Longitudinal Trends in Real-World Outcomes Following Initiation of Ivacaftor: A Cohort Study from the Cystic Fibrosis Registry of Ireland.Ann Am Thorac Soc. 2018 Nov 14. doi: 10.1513/AnnalsATS.201802-149OC. [Epub ahead of print]. [Pubmed]

Fig. 68 Laura Kirwan
LinkedIn
The authors used cystic fibrosis (CF) registry data to assess outcomes following the initiation of ivacaftor, a cystic fibrosis transmembrane conductance regulator (CFTR) potentiator approved for the treatment of CF with a defective gating CFTR mutation.
Longitudinal trends were examined using mixed effects regression analysis in 80 ivacaftor-treated CF patients aged 6-56 years registered with the CF Registry of Ireland with ≥36 month’s pre- and post-commencement data. The effects of ivacaftor treatment on percentage-predicted forced expiratory volume in 1s (ppFEV1), body mass index (BMI), hospitalisation for pulmonary exacerbation, oral and IV antibiotic usage were assessed.
In the 36 months after ivacaftor initiation, ppFEV1 improved by 2.26% per annum [95% CI (0.23 to 4.29), p=0.03] for patients aged <12 years, remained unchanged for 12-<18 years olds [95% CI (-1.87 to 2.88), p=0.67] and declined in adults by 1.74% per annum [95% CI (-3.08 to -0.39), p=0.01]. BMI in adults increased 0.28 kg m-2 per annum [95% CI (0.03 to 0.52), p=0.027] and there was no significant decline in BMI z score in children, [95% CI (-0.01 to 0.13), p=0.08]. In the year after ivacaftor initiation, IV antibiotic treatment reduced by 46% [95% CI (-62.5% to -23.3%), p<0.01], oral antibiotic treatment by 49% [95% CI (-61.1% to -32.1%), p<0.01], and there was no significant reduction in hospitalisation [95% CI (-59.2% to 9.7%), p=0.11].
The authors concluded that in this study of real-world CF registry data, clinical outcomes improved and healthcare resource utilisation reduced after commencing ivacaftor.
Dr Laura Kirwan (fig. 68) is Research Statistician at the Cystic Fibrosis Registry of Ireland. Subsequently, Head of Research and Adjunct Associate Professor University College Dublin.
Kopp BT, McCulloch S, Shrestha CL, Zhang S, Sarzynski L, Woodley FW, Hayes D Jr.Metabolomic responses to lumacaftor/ivacaftor in cystic fibrosis. Pediatr Pulmonol. 2018 May;53(5):583-591. doi: 10.1002/ppul.23972. Epub 2018 Feb 20. [Pubmed]
-

Fig. 69 Benjamin Koop cystic fibrosis news today.com
Lumacaftor/Ivacaftor is a novel CFTR modulator approved for patients with CF that are homozygous for Phe508del CFTR, but its clinical effectiveness varies amongst patients, making it difficult to determine clinical responders. Therefore, identifying biochemical biomarkers associated with drug response are clinically important for follow-up studies.
Serum metabolomics was performed on twenty patients with CF pre- and 6-month post-Lumacaftor/Ivacaftor response via Ultrahigh Performance Liquid Chromatography-Tandem Mass Spectroscopy (UPLC-MS/MS). Correlation with clinical variables was performed. Metabolomics analysis demonstrated 188 differentially regulated metabolites between patients pre- and post-Lumacaftor/Ivacaftor initiation, with a predominance of lipid and amino acid alterations. The top 30 metabolites were able to differentiate pre- and post-Lumacaftor/Ivacaftor status in greater than 90% of patients via a random-forest confusion matrix. Alterations in bile acids, phospholipids, and bacteria-associated metabolites were the predominant changes associated with drug response. Importantly, changes in metabolic patterns were associated with clinical responders.
The authors concluded selected key lipid and amino acid metabolic pathways were significantly affected by Lumacaftor/Ivacaftor initiation and similar pathways were affected in clinical responders. They suggest that targeted metabolomics may provide useful and relevant biomarkers of CFTR modulator responses.
Dr Benjamin Kopp (fig. 69) is a pediatric pulmonologist in Columbus, Ohio and is affiliated with Nationwide Children’s Hospital.
Kourliouros A, Hogg R, Mehew J, Al-Aloul M, Carby M, Lordan JL, Thompson RD, Tsui S, Parmar J. Patient outcomes from time of listing for lung transplantation in the UK: are there disease-specific differences? Thorax. 2018 Oct 3. pii: thoraxjnl-2018-211731. doi: 10.1136/thoraxjnl-2018-211731. [Epub ahead of print][Pubmed]
Data from Department of Cardiac Surgery Royal Brompton. London and the Departments of Cardiothoracic Transplantation at Wythenshawe Manchester, Harefield Hospital, Harefield, Freeman Hospital Newcastle upon Tyne, Queen Elizabeth Hospital Birmingham UK and Royal Papworth, Cambridge and the Department of Statistics and Clinical Studies, NHS Blood and Transplant, Bristol, UK. The authors set out to examine factors influencing patient outcomes from the time of listing for lung transplantation in the UK, examining for differences by patient characteristics, lung disease category and transplant centre.
Data were obtained from the UK Transplant Registry held by NHS Blood and Transplant for adult lung-only registrations between 1January 2004 and 31 March 2014. Pretransplant and post-transplant outcomes were evaluated against lung disease category, blood group and height. Of the 2213 patient registrations, COPD comprised 28.4%, pulmonary fibrosis (PF) 26.2%, cystic fibrosis (CF) 25.4% and other lung pathologies 20.1%. The chance of transplantation after listing differed by the combined effect of disease category and centre (p<0.001). At 3 years postregistration, 78% of patients with COPD were transplanted followed by 61% of patients with CF, 59% of other lung pathology patients and 48% of patients with PF, who also had the highest waiting list mortality (37%). The chance of transplantation also differed by height with taller patients having a greater chance of transplant (HR: 1.03, 95% CI: 1.02 to 1.04, p<0.001). Patients with blood group O had the highest waiting mortality at 3 years post-registration compared with all other blood groups (27% vs 20%, p<0.001).
The authors concluded the way donor lungs were allocated in the UK resulted in discrepancies between the risk profile and probability of lung transplantation. A new donor lung allocation scheme was introduced in 2017 to try to address these shortcomings.
Dr. Antonios Kourilouros is a consultant surgeon in the Department of Cardiac Surgery, Royal Brompton Hospital, London. Subsequently Consultant Cardiac Surgeon at John Radcliffe, Oxford
Norris AW. Is Cystic Fibrosis Related Diabetes Reversible? New Data on CFTR Potentiation and Insulin Secretion. Am J Respir Crit Care Med. 2018 Sep 7. doi: 10.1164/rccm.201808-1501ED. [Epub ahead of print] [Pubmed]
-

Fig. 70 Andrew Norris medicine.uiowa.edu
A discussion on the recent publication of Kelly et el 2018 abstracted above [Pubmed]
- “The Kelly et al result provides hope that restoration of CFTR function might treat and prevent CFRD. However, given the current limited understanding of CFRD pathogenesis, there remains considerable uncertainty. It is possible that the fixed defects in the pancreas might prove too great a burden for the endocrine pancreas to maintain sufficient long term function. Since pancreatic disease begins in utero, it could be envisioned that CFTR restoration may need to begin before birth in order to minimize lifetime CFRD risk. Longer term clinical studies, including in patients with CFRD, will help clarify whether such an intensive approach is needed. Likewise, mechanistic study of the early life determinants of CFRD in relevant CF animal models will help unravel the fixed versus reversible underpinnings of diabetes in
Dr. Andrew Norris is Professor of Biochemistry and Pediatrics – Endocrinology and Diabetes, University of Iowa, Iowa City, Iowa.
Kessler L.Treatment of cystic fibrosis-related diabetes. Lancet Diabetes Endocrinol. 2018 Mar;6(3):167. doi: 10.1016/S2213-8587(18)30042-1. No abstract available. [Pubmed]

Fig. 71 Laurence Kessler
LinkedIn
Laurence Kessler commenting on the recent Ballman et al study (Lancet Diabetes Endocrinol 2018; 6:114-121) comparing the oral anti-diabetic drug repaglinide and insulin for CF related diabetes both of which they found resulted in a similar degree of control. She also mentions a similar previous study by Antoinette Moran et al (Diabetes Care 2009; 32:1783-1788) where those given insulin had an increase in BMI. Kessler notes that the kinetics of regular insulin, with a peak after 3 hours and 6 hour action, does not allow optimum postprandial hyperglycaemia control. He suggest that insulin in the Ballman study may have been under dosed to avoid delayed hypoglaemia – accounting for the absence of weight gain in their insulin group. She mentions the advantage of the newer fast acting insulins (Fiasp, Novo Nordisk) and suggests the availability of the new ultra-fast insulins should enable the design of suitable trials to investigate the effect of treating insulin deficiency in CF related diabetes on metabolism, nutritional and pulmonary outcomes
Laurence Kessler (Fig. 71) is Professor in Endocrinology and Metabolic Disorders, Faculty of Medicine, Strasbourg.
Ballmann M, Holl RW.Treatment of cystic fibrosis-related diabetes – Authors’ reply.Lancet Diabetes Endocrinol. 2018 Mar; 6(3):167-168. doi: 10.1016/S2213-8587(18)30043-3. No abstract available.[Pubmed]
In response to Lawrence Kessler (Lancet Diabetes Endocrinol. 2018 Mar;6(3):167), Manfred Ballman agrees the control of past-prandial hyperglycaemia can be very difficult in CFRD however do not agree their patients were under dosed to avoid hypoglycaemia. He also notes that control was good as judged by the mean HbA1c below 7%. They agree that the newer faster insulins may provide better results but observed that more complicated insulin regimes may further add to problems of long term compliance. Finally, they note, as a reflection of the problems, although Moran et al. in 2009 called for more trials, the Ballman et trial 2018 was the first to be reported since 2009.
Kent DS, Remer T, Blumenthal C, Hunt S, Simonds S, Egert S, Gaskin KJ. 13C-Mixed Triglyceride Breath Test and Fecal Elastase as an Indirect Pancreatic Function Test in Cystic Fibrosis Infants. J Pediatr Gastroenterol Nutr. 2018 Feb 10. doi: 10.1097/MPG.0000000000001905. [Epub ahead of print [Pubmed]
The ‘gold standard’ test for the indirect determination of pancreatic function status in infants with cystic fibrosis (CF), the 72-hour faecal fat excretion test, is likely to become obsolete in the near future. Alternative indirect pancreatic function tests with sufficient sensitivity and specificity to determine pancreatic phenotype need further evaluation in CF infants. Evaluation of the clinical utility of both the non-invasive, non-radioactive C-mixed triglyceride (MTG) breath test and fecal elastase-1 (FE1) in comparison with the 72-hour faecal fat assessment in infants with CF. C MTG breath test and the monoclonal and polyclonal FE1 assessment in stool was compared with the 72-hour faecal fat assessment in 24 infants with CF. Oral pancreatic enzyme substitution (PERT) (if already commenced) was stopped before the tests.
Sensitivity rates between 82-100% for CF patients with pancreatic insufficiency assessed by both the C MTG breath test and the FE1 tests proved to be high and promising. However the C MTG breath test (31-38%), as well as both FE1 tests assessed by the monoclonal (46-54%) and the polyclonal (45%) ELISA-kits showed unacceptably low sensitivity rates for the detection of pancreatic sufficient CF patients in the current study. The authors concluded the C MTG breath test with Non Dispersive Infrared Spectroscopy (NDIRS) technique, as well as both FE1 tests, are not alternatives to the faecal fat balance test for the evaluation of pancreatic function in CF infants during the first year of life.
—- Fecal elastase has proved a very useful non-invasive test when taken with other signs of pancreatic insufficiency even if perhaps less reliable than formal fecal fat balance studies for trials. It is a major advantage that the result is not affected by exogenous pancreatic enzyme supplements.
Dorothea Stark Kent is at the James Fairfax Institute of Paediatric Nutrition, University of Sydney, Sydney, Australia.
Kissner D, LeFlore Y, Narayan SB, Marigowda G, Simard C, Le Camus C False-positive cannabinoid screens in adult cystic fibrosis patients treated with lumacaftor/ivacaftor. J Cyst Fibros. 2018 Sep 11. pii: S1569-1993(18)30754-9. doi: 10.1016/j.jcf.2018.08.006. [Epub ahead of print] Full full text available [Pubmed]
-

Fig. 72 Dana Kissner eMedEvents
It is important that clinicians and patients be aware that false-positive cannabinoid results on urine immunoassay may occur in people taking LUM/IVA, as such results may have negative ramifications for patients in the context of home-, school-, or workplace-related drug screening and eligibility for organ transplantation.
Dr. Dana Kissner (fig.72) is Associate Professor, Internal Medicine at Wayne State University School of Medicine. She was named the 2018 William Stead Tuberculosis Clinician of the Year at the National Tuberculosis Controllers Association annual conference.
Klee P, Dirlewanger M, Lavallard V, McLin VA, Mornand A, Pernin N, Petit LM, Soccal PM, Wildhaber BE, Zumsteg U, Blouin JL, Berney T, Schwitzgebel VM.Combined Pancreatic Islet-Lung-Liver Transplantation in a Pediatric Patient with Cystic Fibrosis-Related Diabetes. Horm Res Paediatr. 2018 Apr 18. doi: 10.1159/000488107. [Epub ahead of print] [Pubmed]

Fig. 73 Philippe Klee
unige.ch
The authors report the first combined pancreatic islet-lung-liver transplantation in a 14-year-old adolescent. The patient was diagnosed with CF at the age of 14 months. Nine years later, after diagnosis of CFRD, the patient’s BMI and lung function began to decline. Bilateral lung transplantation with simultaneous liver transplantation was performed at the age of 14.5 years. The first islet transplantation (IT) was carried out 10 days later. Six months later, C-peptide secretion after arginine stimulation showed peak values of 371 pmol/L (vs. 569 pmol/L before IT) and insulin doses had slightly increased (1.40 vs. 1.11 units/kg/day before IT). A second IT was performed at the age of 15 years, a third at 16 years. Two years after the first IT, arginine-stimulated C-peptide secretion increased to 2,956 pmol/L and insulin doses could be reduced to 0.82 units/kg/day. HbA1c decreased from 7.3% (57.4 mmol/mol) to 5.9% (41.0 mmol/mol).
The authors conclude that islet transplantation following lung and liver transplantation, with injection of islets into a transplanted organ, is feasible. It improves C-peptide secretion, decreases insulin needs, and lowers HbA1c.
Philippe Klee (fig. 73) is physician at the Pediatric Endocrine and Diabetes Unit, Department of Pediatrics, University Hospitals of Geneva, Geneva, Switzerland and the Diabetes Center of the Faculty of Medicine, University of Geneva, Geneva, Switzerland.
Kim SO, Corey M, Stephenson AL, Strug LJ. Reference percentiles of FEV1 for the Canadian cystic fibrosis population: comparisons across time and countries.Thorax. 2018 Feb 6. pii: thoraxjnl-2017-210899. doi: 10.1136/thoraxjnl-2017-210899. [Epub ahead of print][Pubmed]

Fig. 73 Sang Ook Kim
LinkedIn
Forced expiratory volume in 1 s (FEV1) indicates lung health in cystic fibrosis (CF). FEV1 is commonly communicated as a per cent predicted of a healthy individual sharing the same age, sex, race and height. CF-specific reference equations are complementary and calibrate a patient’s FEV1 to that of their CF peers. Objectives were
(1) To derive Canadian CF-specific FEV1 reference percentiles (FEV1%iles), (2) characterize how they have changed over time and (3) compare the Canadian FEV1%iles to those for USA and European CF populations. CF FEV1%iles are calculated using the Canadian CF Registry and quantile regression. The Canadian FEV1%iles demonstrated better lung function in more recent time periods within Canada, especially below the 50% percentile and in males. When compared to USA and European FEV1%iles for the same time period, Canadian FEV1%iles were higher. The authors concluded CF-specific FEV1%iles can provide useful information about changes in lung health. An online calculator (available at cfpercentilesickkids.ca) makes these FEV1%iles accessible.
Dr Sang Ook Kim (fig.73) is in the Division of Biostatistics, Dalla Lana School of Public Health, University of Toronto, Toronto, Canada. Research Institute, The Hospital for Sick Children, Toronto, Canada.
Keogh RH, Szczesniak R, Taylor-Robinson D, Bilton D. Up-to-date and projected estimates of survival for people with cystic fibrosis using baseline characteristics: A longitudinal study using UK patient registry data. J Cyst Fibros. 2018 Jan 5. pii: S1569-1993(17)30976-1. doi: 10.1016/j.jcf.2017.11.019. [Epub ahead of print] Free full text[Pubmed]
Flexible parametric survival modelling of UK CF Registry data from 2011 to 2015, capturing 602 deaths in 10,428 individuals. Survival curves were estimated from birth; conditional on reaching older ages; and projected under different assumptions concerning future mortality trends, using baseline characteristics of sex, CFTR genotype (zero, one, two copies of F508del) and age at diagnosis. Male sex was associated with better survival, as was older age at diagnosis, but only in F508del non-homozygotes. Survival did not differ by genotype among individuals diagnosed at birth. Median survival ages at birth in F508del homozygotes were 46years (males) and 41years (females), and similar in non-homozygotes diagnosed at birth. F508del heterozygotes diagnosed aged 5 had median survival ages of 57 (males) and 51 (females). Conditional on survival to 30, median survival age rises to 52 (males) and 49 (females) in homozygotes. Mortality rates decreased annually by 2% during 2006-2015. Future improvements at this rate suggest median survival ages for F508del homozygous babies of 65 (males) and 56 (females). Over half of babies born today, and of individuals aged 30 and above today, can expect to survive into at least their fifth decade.
— Important and encouraging information from the CF Trust’s patient registry. The lead author Dr Ruth Keogh of the London School of Hygiene and Tropical Medicine also has an explanatory article on the meaning of the findings on the CF Trust website. “What is median survival age?”
-

Fig. 74 Ruth Keogh
Dr Ruth Keogh is Statistician and Associate Professor at the London School of Hygiene and Tropical Medicine. She joined the Medical Statistics Department at LSHTM in 2012. Previously she studied Mathematics and Statistics at the University of Edinburgh, an MSc in Applied Statistics at the University of Oxford, and a DPhil in Medical Statistics/Epidemiology also in Oxford. Prior to joining LSHTM she worked at the MRC Biostatistics Unit, Cambridge (2008-2012) and the Cancer Epidemiology Unit, University of Oxford (2006-2007).
— It is good that the major efforts to develop the CF Trust UK patient registry, starting back in the Eighties by Anil and Gita Mehta in Dundee and the eventually combining with the CF Foundation system, are being subjected to such professional analysis with results that are available to families and people with CF in an understandable form.
Kirby T. Tezacaftor-ivacaftor is safe and efficacious in patients with cystic fibrosis with Phe508del mutations. Lancet Respir Med. 2018 Jan;6(1):13-14. doi: 10.1016/S2213-2600(17)30439-3. Epub 2017 Dec 14. [Pubmed]
Two phase 3 double blind, randomised, placebo-controlled trials published in the New England Journal of Medicine show that treatment with the combination of tezacaftor-ivacaftor in patients with cystic fibrosis aged 12 years and older who have the common cystic fibrosis transmembrane conductance regulator ( CFTR ) gene mutation, Phe508del, is both safe and efficacious.
— This is a useful brief update by a medical journalist on the present trials and position regarding the recent tezacaftor-ivacaftor combinations with mention of encouraging developments in the triple combination therapy.
— For details of the trial in Phe508del heterozygotes see Rowe et al. N Eng J Med 2017; 377:2024-35 ([Pubmed]); for that in Phe508del homozygotes see Taylor-Cousar et al. N Eng J Med 2017; 377:2013-23 ([Pubmed] both abstracted in the 2017 section of this History.
Konstan MW, Van Devanter DR, Sawicki GS, Pasta DJ, Foreman AJ, Neiman EA, Morgan WJ. Association of High-Dose Ibuprofen Use, Lung Function Decline, and Long-Term Survival in Children with Cystic Fibrosis.Ann Am Thorac Soc. 2018 Jan 9. doi: 10.1513/AnnalsATS.201706-486OC. [Epub ahead of print] [Pubmed]
-

Fig. 75 Michael Konstan
A study of associations of high-dose ibuprofen treatment with the rate of FEV1 decline and mortality among children followed in the Epidemiologic Study of Cystic Fibrosis (ESCF) and subsequently in the US Cystic Fibrosis Foundation Patient Registry (CFFPR). In a propensity-score matched cohort study of children with CF, the authors observed an association between high-dose ibuprofen use and both slower lung function decline and improved long-term survival. They suggest these results are consistent with the hypothesis that treatment-associated reduction of lung function decline in children with CF leads to improved survival-
— Despite the importance of inflammation in the causation of lung damage and the previous work on ibuprofen for people with CF by the leading author and others, ibuprofen is prescribed for very few patients and did not figure in the prescribed recommended therapies in the 2015 CFF patient registry; also less than 2 per cent of adults were taking ibuprofen treatment.
Korten I, Kieninger E, Yammine S, Cangiano G, Nyilas S, Anagnostopoulou P, Singer F, Kuehni CE, Regamey N, Frey U, Casaulta C, Spycher BD, Latzin P; SCILD; BILD study group Respiratory rate in infants with cystic fibrosis throughout the first year of life and association with lung clearance index measured shortly after birth. J Cyst Fibros. 2018 Jul 27. pii: S1569-1993(18)30696-9. doi: 10.1016/j.jcf.2018.07.002. [Epub ahead of print] [Pubmed]
-

Fig. 76 Philippe Latzin
- Lung impairment in cystic fibrosis (CF) starts in infancy. However,tools to monitor early lung disease are limited. Respiratory rate (RR) as a key vital sign is easy to assess during sleep and is elevated during acute respiratory disease. Thus, elevated RR could indicate early lung impairment and potentially serve as a diagnostic tool in disease monitoring. In a prospective cohort of infants with CF diagnosed by newborn screening and healthy controls RR was measured and respiratory symptoms reported weekly throughout infancy. Infants performed a lung function measurement within the first weeks of life. The analyses included 5656 measurements from 153 infants (43 with CF). RR declined from 43.2 (40.5)/min at 6 weeks of age to 28.3 (24.6)/min at 50 weeks in infants with CF (healthy controls). Infants with CF had consistently higher RR than controls (mean difference: 4.15/min; (95% CI 2.86-5.44); p < .001). In both study groups, RR was increased throughout the study period in infants with higher lung clearance indices (LCI) and during episodes of respiratory infections.
The authors concluded infants with CF have a higher RR compared to healthy controls during the first year of life. The association with early LCI measurements, the current gold standard to assess physiology of peripheral airways, persisted throughout the study period. This may indicate tracking of lung function by RR. It might thus be an early subtle sign of functional respiratory deficit. Further studies will show if RR can be used as a sensitive and promising marker to monitor early CF lung disease.
The corresponding author, Philippe Latzin (fig.76) is Head of Paediatric Pulmonology at University of Berne Children’s Hospital.
Kounis I, Lévy P, Rebours V.Ivacaftor CFTR Potentiator Therapy is Efficient for Pancreatic Manifestations in Cystic Fibrosis. Am J Gastroenterol. 2018 Jul;113(7):1058-1059. doi: 10.1038/s41395-018-0123-7. Epub 2018 Jun 11. [Pubmed]
No abstract
Labonte ML. The Mist Tent: An Analysis of Therapeutic Change in the History of Cystic Fibrosis Care.Bull Hist Med. 2018;92(4):634-663. doi: 10.1353/bhm.2018.0074. [PubMed]

Fig. 77 Michelle LaBonte
Harvard University
Mist tent therapy for cystic fibrosis went through a rise and fall in popularity between the 1950s and 1970s, providing an opportunity to explore the nature of therapeutic change in medicine. The therapy “worked” in the context of a particularly grim life expectancy in the early 1950s and in the setting of a comprehensive therapeutic program that began in Cleveland in 1957. Although clinical studies published in the 1970s provided evidence that mist tents were ineffective or even harmful, these later studies were not necessarily more robust than earlier studies that provided evidence of mist tent efficacy, suggesting that other factors may have also contributed to mist tent abandonment. In fact, the unpalatable nature of mist tent therapy, which was described by one doctor as akin to incarceration, and studies that questioned the theoretical underpinnings of the therapy also played important roles in the eventual abandonment of misttents.
– As noted above, before dismissing the use of mist tents it is worth considering the increasing support for the importance of the fluid layer of the respiratory epithelium and also Robert Wood’s (1976 above [PubMed]) comments that he had observed considerable moisture in the airways when bronchoscoping patients after mist tent therapy! The present article provides a very detailed and readable account of the mist tent therapy over the years. See also Topic section of this History.
Michelle LaBonte (fig. 77) is Assistant Professor of History, Dept of History of Science, Harvard University
Laselva O, Molinski S, Casavola V Bear CE. Correctors of the major Cystic Fibrosis mutant interact through membrane spanning domains. Mol Pharmacol. 2018 Apr 4. pii: mol.118.111799. doi:10.1124/mol.118.111799. [Epub ahead of print]Full text [Pubmed]

Fig. 78 Onofrio Laselva ResearchGate
The most common cystic fibrosis causing mutation is deletion of phenylalanine 508 (F508del), a mutation that leads to protein misassembly with defective processing. Small molecule correctors compounds: VX-809 or Corr-4a (C4) partially restore processing of the major mutant. These two prototypical corrector compounds cause an additive effect on F508del-CFTR processing and hence were proposed to act through distinct mechanisms: VX-809 stabilizing the first membrane spanning domain 1 (MSD1) and C4, acting on the second half of the molecule (consisting of MSD2 and/or nucleotide binding domain 2 (NBD2)). The authors confirmed the effect of VX-809 in enhancing the stability of MSD1 and showed that it also allosterically modulates MSD2
when co-expressed with MSD1. They showed for the first time that C4 stabilizes the second half of the CFTR protein through its action on MSD2. Given the allosteric effect of VX-809 on MSD2- they were prompted to test the hypothesis that the two correctors interact in the full length mutant protein. They did see evidence supporting their interaction in the full-length F508del-CFTR protein bearing secondary mutations targeting domain:domain interfaces. Disruption of the MSD1:F508del-NBD1 interaction (R170G) prevented correction by both compounds pointing to the importance of this interface in processing. On the other hand, stabilization of the MSD2: F508del-NBD1 interface (by introducing R1070W) led to a synergistic effect of the compound combination on the total abundance of both the immature and mature form of the protein. Together- these findings suggest that the two correctors interact in stabilizing the complex of MSDs in F508del-CFTR.
Dr. Onofrio Laselva (fig.78) is a post doctoral Research Scientist at the Hospital for Sick Children Toronto.
– The full text is available for any scientists who require more detail.
LeGrys VA, Moon TC, Laux J, Rock MJ, Accurso F. Analytical and biological variation in repeated sweat chloride concentrations in clinical trials for CFTR modulator therapy. J Cyst Fibros. 2018 Jan;17(1):43-49. doi: 10.1016/j.jcf.2017.07.008. Epub 2017 Jul 22 [Pubmed]

Fig. 79 Vicky A Le Grys
med.unc.edu
Using sweat chloride as a biomarker for CFTR modifying drugs requires knowledge of analytical and biological variation. 979 sweat chloride concentrations from 128 subjects enrolled in the placebo arm of 2 multicentre, investigational drug trials were analysed to determine coefficients of variation (CV) as well as reference change value (RCV) and index of individuality (II).
- For these populations, calculated values for the two studies were: analytical variation (3.9, 4.1%); within-subject variation (4.4, 6.0%); between-subject variation (8.9, 7.0%); RCV (13.7, 17.0%) and II (0.7, 1.0). Sweat chloride variation was not affected by sex, collection site or sample weight; but was slightly affected by age in one of the two studies.
Through determination of analytical as well as between- and within-subject variation, and with a larger sample size, our data allows improved estimates of the RCV and II, and can contribute to future trials of CFTR modulators and inform the design and interpretation of n of 1 trials in both research and clinical settings.
Dr. Vicky A LeGrys (fig. 79) is a scientist and Professor in the Department of Allied Sciences, University of North Carolina. She is an expert on all aspects of sweat testing.
Lee SY, Chesdachai S, Lee MJ, He XM, Tangpricha V, Braverman LE.Thyroid Function in Patients with Cystic Fibrosis: No Longer a Concern? Thyroid. 2016 Jul;26(7):875-9. doi: 10.1089/thy.2015.0567. Epub 2016 May 19. [Pubmed]
-

Fig. 80 Sun Y Lee US News Health
- Development of goitre and hypothyroidism has been reported in patients with cystic fibrosis (CF) since the 1970s, especially when treated with iodine-based expectorants. With iodine-containing expectorants no longer in routine use, the prevalence of thyroid dysfunction in CF patients is unknown. This cross-sectional study assessed thyroid function status in a large cohort of CF patients.
Of the 87 subjects with measured TSH values, seven (8%) had abnormal levels (range 0.2-7.6 μIU/mL; one overt, four subclinical hypothyroidism, and two subclinical hyperthyroidism). Of the 56 subjects with measured fT4 values, 19 (34%) had slightly low levels (range 0.49-0.79 ng/dL; 17 isolated mild hypothyroxinemia). A positive correlation between age and body mass index (BMI; p < 0.001) and a negative correlation between age and FEV1 (p = 0.041) were seen. Age, sex, race/ethnicity, BMI, FEV1, hospitalization status, use of pancreatic enzyme or thyroid hormone replacement, recent antibiotic use, and TPO antibody positivity were not predictive of TSH, fT4, or thyroid dysfunction risk. Stratified analyses by hospitalization did not predict TSH or fT4.
The authors concluded that although 24 (27%) of the patients had abnormal serum thyroid function tests, overt thyroid dysfunction was rare in this cohort of 89 patients with CF. The degree of hypothyroxinemia was marginal, likely due to non-thyroidal illness. There were no significant predictors of thyroid dysfunction.
Dr. Sun Y Lee (fig 80) is Assistant Professor, Boston University School of Medicine, endocrinology, diabetes and nutrition.
— Previous work on thyroid function in CF, including the relation to iodide respiratory therapy, has been reviewed in the Topics – Endocrinology section of this website (cysticfibrosis.online)
Lehoux Dubois C, Labrèche E, Boudreau V, Colomba J, Mailhot M, Lavoie A, Rabasa-Lhoret R, Coriati A. Extra-skeletal impact of vitamin D supplementation protocol in an adult population with cystic fibrosis.Clin Nutr. 2018 Aug 25. pii: S0261-5614(18)32384-7. doi: 10.1016/j.clnu.2018.08.013. [Epub ahead of print] [Pubmed]
-

Fig. 81 Catherine Lehoux Dubois ResearchGate
- The aim was to study the association between serum vitamin D levels and key clinical factors, such as nutritional status, pulmonary function and pulmonary exacerbations (PEx) frequency, in an adult CF population. Prospective analysis of a published vitamin D (VitD3) supplementation protocol (N = 200 adult patients) over a follow-up period of 5 years. Data were collected from the medical files before (baseline) and after (follow-up) the implementation of the VitD3 supplementation protocol, between 2009 and 2014. Serum samples to measure vitamin D were also collected at baseline and follow-up.
A positive relationship between serum vitamin D and lung function was observed at baseline (R = 0.158, P = 0.027), but it disappeared at follow-up (P = 0.454). There was no association between serum vitamin D levels and body mass index. At follow-up, patients with significantly higher serum vitamin D levels were women, older in age, had CF-related diabetes or had a history of recurring PEx.
The authors concluded there was no direct link between heightened serum vitamin D and lung function or BMI in an adult CF population. They suggest that better compliance to treatments and closer follow-up from health professionals could partially explain why such patients reached higher vitamin D serum levels.
Catherine Lehoux Dubois (fig. 81) is based at the Institut de recherches clinicque de Mantreal, Canada
McCormick J, Cho DY, Lampkin B, Richman J, Hathorne H, Rowe SM, Woodworth BA. Ivacaftor improves rhinologic, psychologic, and sleep-related quality of life in G551D cystic fibrosis patients. Int Forum Allergy Rhinol. 2018 Nov 24. doi: 10.1002/alr.22251. [Epub ahead of print] [PubMed].

Fig. 82 Justin McCormick
LinkedIn.com
The G551D Observational (GOAL) study was a multi-center prospective cohort study enrolling CF patients ≥6 years with at least 1 G551D mutation. Subjects were provided 20-item Sino-Nasal Outcome Test (SNOT-20) questionnaires prior to ivacaftor therapy and at 1, 3, and 6 months afterward. The impact on rhinologic (R), psychological (P), sleep (S), and ear/facial (E) quality of life (QOL) domains was evaluated separately.
Of 153 subjects, 129 (84%) completed all questionnaires. Ivacaftor improves QOL in the R, P, and S domains in G551D CF patients, although QOL instruments validated for CRS may not translate well to CF CRS patients because symptom burden was surprisingly low.
Justin McCormick is in the Department of Otolaryngology-Head and Neck Surgery, University of Alabama at Birmingham, Birmingham Alabama.
Millar BC, Rendall JC, Downey DG, Moore JE. Does ivacaftor interfere with the antimicrobial activity of commonly used antibiotics against Pseudomonas aeruginosa?-Results of an in vitro study. J Clin Pharm Ther. 2018 Dec;43(6):836-843. doi: 10.1111/jcpt.12722. Epub 2018 Jun 29.[Pubmed]

Fig. 83 Beverley Cherie Millar
LinkedIn
Little is known about the potential effects of ivacaftor on bacterial ion channels, which, in turn, may have a potential effect on transport across the bacterial cell membrane. Therefore, any change in the ability to transport molecules across cell membranes in bacteria could have an important impact on bacterial transport physiology. One area where this could be particularly important is in the movement of antibiotics, both into and out of the bacterial cell. An in vitro study was therefore performed to examine the influence of ivacaftor at therapeutic concentration on antibiotic susceptibility of 11 commonly used anti-pseudomonal antibiotics against a population of clinical Pseudomonas aeruginosa [PA], from CF and non-CF sources.
Antibiotic susceptibility did not decrease significantly with any of the antibiotics examined with CF PA isolates or with non-CF PA control organisms. There was a statistically significant increase in zone size (CF PA and amikacin, gentamicin, temocillin and ciprofloxacin; Non-CF PA and amikacin, gentamicin and aztreonam). However, at a population level, this did not translate into a shift in CLSI category to a more susceptible phenotype. None of the PA isolates examined were susceptible to ivacaftor alone, and additionally, no changes were noted with the four phenotypic parameters examined in the presence of ivacaftor.
— This study is reassuring and showed that antibiotic susceptibility of commonly used anti-pseudomonal antibiotics was not negatively affected by ivacaftor, in a population of ivacaftor-naive P. aeruginosa.
Beverley Cherie Millar (fig.83) is a clinical scientist based at the North of Ireland Public Health Laboratory, Department of Bacteriology, Belfast City Hospital, Belfast UK.and the School of Biomedical Sciences, Ulster University, Coleraine, UK.
Murer C, Huber LC, Kurowski T, Hirt A1, Robinson CA, Bürgi U, Benden C. First experience in Switzerland in Phe508del homozygous cystic fibrosis patients with end-stage pulmonary disease enrolled in a lumacaftor-ivacaftor therapy trial – preliminary results. Swiss Med Wkly. 2018 Feb 16;148:w14593. doi: 10.4414/smw.2018.14593. eCollection 2018.Free full text[Pubmed]

Fig. 84 Christian Benden
ResearchGate
Patients with FEV1 <40% predicted were excluded from previous studies. The authors used LUM/IVA on a “compassionate use” basis in cystic fibrosis patients with end-stage pulmonary disease. All patients from the Adult Cystic Fibrosis Centre at the University Hospital Zurich were included who were Phe508del homozygous genotype and a predicted FEV1 <40% or being evaluated or already listed for LTX.
Twenty patients were on trial with LUM/IVA; at the cut-off date, 6-month follow-up was complete for 10 patients. Respiratory related adverse events (RAEs) were severe and occurred early. The dropout rate due to RAE or lack of clinical success was 20%. Median acute exacerbation rate (AER) decreased from 2.5 in the 6 months pre-treatment to 1 during the observation period. FEV1 increased from 32 to 34.5% predicted, p = 0.292. The 6-MWD increased by a median 33 m (p = 0.6086). Sweat chloride decreased significantly by a median of 25 mmol/l (p = 0.0003). Median BMI increased from 19 to 19.9 kg/m2 (p = 0.1488). At the cut-off, three previously listed patients were paused on the transplant waiting list.
The authors concluded Phe508del homozygous cystic fibrosis patients with end-stage pulmonary disease tolerated LUM/IVA, although respiratory related adverse events occurred early and were severe. This positive finding was probably due to the stepwise dose increases. There was clinical benefit mainly from reduction in acute exacerbation rate and stabilisation of lung function. They propose that all suitable Phe508del homozygous cystic fibrosis patients with end-stage pulmonary disease should have a trial of LUM/IVA treatment in experienced centres.
Dr Christian Benden (fig.84) is Medical Director Lung Transplantation and CF Center Director, Division of Pulmonology, University Hospital Zurich, Switzerland
Popowicz N, Wood J, Tai A, Morey S, Mulrennan S .Immediate effects of lumacaftor/ivacaftor administration on lung function in patients with severe cystic fibrosis lung disease. J Cyst Fibros. 2017 May;16(3):392-394. doi: 10.1016/j.jcf.2017.02.009. Epub 2017 Mar 15. [Pubmed]

Fig 85. Natalia Popowicz
ResearchGate
Safety-data for lumacaftor/ivacaftor (LUM/IVA) combination therapy in patients with severe lung disease (percent predicted forced expiratory volume in 1s [ppFEV1] <40) remain limited.
The authors report immediate post-dose respiratory-related adverse events in 12 patients with severe cystic fibrosis (CF) lung disease (median [IQR] ppFEV1: 34 [31-36]) prescribed LUM/IVA. All patients experienced a decline in ppFEV1 from baseline at 2-hours (median [IQR] relative change: -19 [-21 to -11]%, p<0.001) that persisted at 24-hours but recovered in most patients at 1-month. No pre- and post-differences in bronchodilator response were observed. Ten (83.3%) patients reported non-severe respiratory-related adverse events within 24-hours of LUM/IVA initiation. At 1-month, eight (67%) patients had persistent symptoms and six (50%) were treated for a pulmonary exacerbation.
The authors suggest their results highlight that LUM/IVA respiratory-related adverse events are common in patients with a ppFEV1<40. They recommend close assessment of adverse events. Further studies are required to evaluate the efficacy of LUM/IVA in patients with severe lung disease.
Natalia Popowicz (fig.85) is Senior Clinical Pharmacist in the Pharmacy Department, Sir Charles Gairdner Hospital, Perth, Australia; Respiratory Medicine, Sir Charles Gairdner Hospital, Perth, Australia; School of Medicine & Pharmacology, University of Western Australia, Perth, Australia
Li H,Pesce E,Sheppard DN,Singh AK,Pedemonte N.Therapeutic approaches to CFTR dysfunction: From discovery to drug development. J Cyst Fibros.2018Mar;17(2S):S14-S21. doi: 10.1016/j.jcf.2017.08.013. Epub 2017 Sep 12 [Pubmed]
Cystic fibrosis (CF) mutations have complex effects on the cystic fibrosis transmembrane conductance regulator (CFTR) protein. They disrupt its processing to and stability at the plasma membrane and function as an ATP-gated Cl- channel. Here, the authors review therapeutic strategies to overcome defective CFTR processing and stability. Because CF mutations have multiple impacts on the assembly of CFTR protein, combination therapy with several pharmacological chaperones is likely to be required to rescue mutant CFTR expression at the plasma membrane. Alternatively, proteostasis regulators, proteins that regulate the synthesis, intracellular transport and membrane stability of CFTR might be targeted to enhance the plasma membrane expression of mutant CFTR. Finally, they consider an innovative approach to bypass CFTR dysfunction in CF, the delivery of artificial anion transporters to CF epithelia to shuttle Cl- across the apical membrane. The identification of therapies or combinations of therapies, which rescue all CF mutations, is now a priority.
– A neat summary of the present situation from Professor David Sheppard’s team in Bristol.
Hongyu Li is Senior Research Fellow at the School of Physiology, Pharmacology and Neuroscience, University of Bristol, Biomedical Sciences Building, University Walk, Bristol BS8 1TD, United Kingdom.
Mannik LA, Chang KA, Annoh PQK, Sykes J, Gilmour J, Robert R, Stephenson AL. Prevalence of hypoglycemia during oral glucose tolerance testing in adults with cystic fibrosis and risk of developing cystic fibrosis-related diabetes. J Cyst Fibros. 2018 Apr 18. pii: S1569-1993(18)30081-X. doi: 10.1016/j.jcf.2018.03.009. [Epub ahead of print] [Pubmed]

Fig. 86 Lisa Mannik
LinkedIn
Few studies have examined whether hypoglycemia during the OGTT increases the risk of developing CF-related diabetes (CFRD). Objectives of this study were to describe the characteristics of CF patients with hypoglycemia during the OGTT and to determine the incidence and time to development of CFRD in those with hypoglycaemia. 138 patients (29.6%) experienced hypoglycaemia during the OGTT. More males experienced hypoglycaemia compared to no hypoglycemia (69.6% vs. 54.6% respectively; p = 0.003). Those who were heterozygous deltaF508 were more likely to experience hypoglycemia (p = 0.006). Subjects who experienced hypoglycemia were less likely to develop CFRD at ten years compared to no hypoglycemia (12.0% vs. 42.1%, respectively; p < 0.001). Hypoglycemia following OGTT is common in CF however the 10 year risk of developing CFRD in these patients was low. Males and those who were heterozygous deltaF508 were at higher risk for hypoglycemia.
Lisa A Mannik is Registered Dietitian in the Department of Respirology, St. Michael’s Hospital, 30 Bond Street, Toronto, Ontario M5B 1W8, Canada.
Mayer-Hamblett N, Retsch-Bogart G, Kloster M, Accurso F, Rosenfeld M, Albers G, Black P, Brown P, Cairns A, Davis SD, Graff GR, Kerby GS, Orenstein D, Buckingham R, Ramsey BW; OPTIMIZE Study Group. Azithromycin for Early Pseudomonas Infection in Cystic Fibrosis: The Optimize Randomized Trial. Am J Respir Crit Care Med. 2018 Jun 11. doi: 10.1164/rccm.201802-0215OC. [Epub ahead of print] [Pubmed]
-

Fig. 87 Nicole Mayer-Hamblett Cystic Fibrosis Foundation
A study to test the hypothesis that the addition of azithromycin to TIS in children with CF and early Pa decreases the risk of PEx and prolongs time to Pa recurrence.The OPTIMIZE trial was a multicenter, double-blind, randomized, placebo-controlled, 18-month trial in children with CF ages 6 months-18 years with early Pa. Azithromycin or placebo was given 3x weekly with standardized TIS. The primary endpoint was time to PEx requiring antibiotics and secondary endpoint time to Pa recurrence, in addition to other clinical and safety outcomes.
221 participants (111 placebo, 110 azithromycin) out of a planned 274 were enrolled. Enrolment was stopped early by the National Heart, Lung, and Blood Institute (NHLBI) because the trial had reached the pre-specified interim boundary for efficacy. The risk of PEx was reduced by 44% in the azithromycin group as compared to placebo (hazard ratio [HR]: 0.56, 95% CI:0.37,0.83, p=0.004). Weight increased by 1.27 kg in the azithromycin group compared to placebo (95% CI:0.01,2.52, p=0.046). No significant differences were seen in microbiologic, clinical, or safety endpoints.
The authors concluded azithromycin was associated with a significant reduction in risk of PEx and sustained improvement in weight but had no impact on microbiologic outcomes in children with early Pa.
Nicole Mayer-Hamblett (fig. 87) is an Associate Professor in the Department of Pediatrics and Adjunct Associate Professor in the Department of Biostatistics at the University of Washington.
Masson A, Schneider-Futschik EK, Baatallah N, Nguyen-Khoa T, Girodon E, Hatton A, Flament T, Le Bourgeois M, Chedevergne F, Bailly C, Kyrilli S, Achimastos D, Hinzpeter A, Edelman A, Sermet-Gaudelus I. Predictive factors for lumacaftor/ivacaftor clinical response. J Cyst Fibros. 2019 May;18(3):368-374. doi: 10.1016/j.jcf.2018.12.011. Epub 2018 Dec 28 [Pubmed]
Ivacaftor-lumacaftor combination therapy corrects the F508 del-CFTR mutated protein which causes Cystic Fibrosis. The clinical response of the patients treated with the combination therapy is highly variable. This study aimed to determine factors involved in the individual’s response to lumacaftor-ivacaftor therapy.
Sweat test was assessed at baseline and after 6 months of ivacaftor-lumacaftor treatment in 41 homozygous F508del children and young adults. β-adrenergic peak sweat secretion, nasal potential difference (NPD) and intestinal current measurements (ICM) were performed in patients accepting these tests. Serum level of lumacaftor and ivacaftor were determined and additional CFTR variant were searched.
Results: Sweat chloride concentration significantly decreased after treatment, whereas the β-adrenergic peak sweat response did not vary in 9 patients who underwent these tests. The average level of F508del-CFTR activity rescue reached up to 15% of the normal level in intestinal epithelium, as studied by ICM in 12 patients (p = .03) and 20% of normal in the nasal epithelium in NPD tests performed in 21 patients (NS). There was no significant correlation between these changes and improvements in FEV1 at 6 months. Serum drug levels did not correlate with changes in FEV1, BMI-Z score or other CFTR activity biomarkers. Additional exonic variants were identified in 4 patients. The F87L-I1027T-F508del-CFTR complex allele abolished the lumacaftor corrector effect.
Conclusion: This observational study investigates a number of potential factors linked to the clinical response of F508del homozygous patients treated with lumacaftor-ivacaftor combination therapy. Lumacaftor and ivacaftor blood levels are not associated with the clinical response. Additional exonic variants may influence protein correction.
Alexandra Masson is at the Centre Maladie Rare Mucoviscidose, Hôpital Necker-Enfants Malades, Assistance-Publique Hôpitaux de Paris
McColley SA, Konstan MW, Ramsey BW, Stuart Elborn J, Boyle MP, Wainwright CE, Waltz D. Vera-Llonch M, Marigowda G, Jiang JG, Rubin JL.Lumacaftor/Ivacaftor reduces pulmonary exacerbations in patients irrespective of initial changes in FEV1. J Cyst Fibros. 2018 Aug 23. pii: S1569-1993(18)30719-7. doi: 10.1016/j.jcf.2018.07.011. [Epub ahead of print] hnuFree full text[Pubmed]

Fig.88 Susanna McColley
Lurie Children’s
Improved lung function and fewer pulmonary exacerbations (PEx) were observed with lumacaftor/ivacaftor (LUM/IVA) in patients with cystic fibrosis homozygous for F508del. It is unknown whether PEx reduction extends to patients without early lung function improvement.
Post hoc analyses of pooled phase 3 data (NCT01807923, NCT01807949) categorized LUM/IVA-treated patients by per cent predicted forced expiratory volume in 1 s (ppFEV1) change from baseline to day 15 into threshold categories (absolute change ≤0 vs >0; relative change <5% vs ≥5%) and compared PEx rates vs placebo. LUM (400 mg q12h)/IVA (250 mg q12h)-treated patients (n = 369) experienced significantly fewer PEx vs placebo, regardless of threshold category. With LUM/IVA, PEx rate per patient per year was 0.60 for those with absolute change in ppFEV1 > 0 and 0.85 for those with absolute change ≤0 (respective rate ratios vs placebo [95% CI]: 0.53 [0.40-0.69; P < .0001], 0.74 [0.55-0.99; P = .04]).
The authors concluded LUM/IVA significantly reduced PEx, even in patients without early lung function improvement.
Susanna McCauley (fig.88) is at the Ann & Robert H. Lurie Children’s Hospital of Chicago and Northwestern University Feinberg School of Medicine, 225 East Chicago Avenue #43, Chicago, IL 60611, USA.
Meynet E, Laurin D, Lenormand JL, Camara B, Toussaint B, Le Gouëllec A. Killed but metabolically active Pseudomonas aeruginosa-based vaccine induces protective humoral- and cell-mediated immunity against Pseudomonas aeruginosa pulmonary infections. Vaccine. 2018 Mar 27;36(14):1893-1900. doi: 10.1016/j.vaccine.2018.02.040. Epub 2018 Mar 2. [Pubmed]
In this study, the authors developed an innovative and safer live-attenuated Pa vaccine based on a Killed But Metabolically Active (KBMA) attenuation method. KBMA Pa has been further rationally designed to overexpress beneficial effectors like the type 3 secretion system apparatus. They demonstrated that KBMA Pa elicits a high and broad humoral response in mice against several antigens of particular interest such as OprF and PcrV proteins. Moreover, they assessed cytokines in the serum of immunized mice and showed that KBMA Pa elicits Th1, Th2 and especially Th17 pathways of cell-mediated immune responses. Th17 pathway involvement was also confirmed after specific stimulation of helper T cells in immunized mice. Finally, they showed that this vaccine is safe and has a protective effect in a murine acute pulmonary infectious challenge. In conclusion, they consider that KBMA Pa is a new platform with high potential for the development of a vaccine against Pa.
— All previous vaccines against P. aeruginosa have been ineffective including one of the most promising, that of Dr. A B Lang and colleagues (Lang AB et al, Pediatr Infect Dis 2004; 23:504-510) where the apparently impressive results of their preliminary study prompted an international multicentre trial of the vaccine. Unfortunately the results of this major trial were entirely negative – there being no difference between the treated and control groups either in the prevalence of chronic P. aeruginosa infection or the number of new positive Pseudomonas respiratory cultures. It was possible that the increasing use of anti-Pseudomonas eradication therapy and the fall in the prevalence of chronic Pseudomonas infection in clinics (and the opportunity to become infected) contributed to the negative results (unpublished data).
— There was still no vaccine for P.aeruginosa by 2024.
Millar BC, McCaughan J, Rendall JC, Downey DG, Moore JE. Pseudomonas aeruginosa in cystic fibrosis patients with c.1652G›A (G551D)-CFTR treated with ivacaftor-Changes in microbiological parameters. J Clin Pharm Ther. 2018 Feb;43(1):92-100. doi: 10.1111/jcpt.12616. Epub 2017 Sep 22. [Pubmed]
This study shows that microbiological parameters with patients on ivacaftor (IVA) therapy were not detrimentally affected, as there was a lower requirement IV antibiotic therapy and a reduction in the rate and density of mucoid Pseudomonas aeruginosa (M‐PA). Examination of antibiotic susceptibility, as expressed by Relative Resistance Index, against four classes of antibiotic in (a) total PA, (b) Non‐mucoid PA (NM‐PA) and (c) M‐PA, isolated from patients before and after commencement of ivacaftor therapy, showed no reduction in antibiotic susceptibility. Overall, this study suggests an improvement in the microbiology status of patients on IVA therapy,
Following commencement of IVA therapy, patients required less iv antibiotic courses but no change in number of oral antibiotics courses. There was significant reduction in both the rate of isolation and density of M-PA (P = .02; P = .006, respectively). In contrast, there was no significant reduction in both the rate of isolation and density of NM-PA (P = .90; P = .07, respectively). Antimicrobial susceptibility in NM-PA and M-PA was not significantly reduced within any of the antibiotics classes or individual antibiotics examined. Increased susceptibility was noted in the beta-lactam class for NM-PA and M-PA, in particular with ceftazidime.Overall, (i) the requirement for less iv antibiotic therapy,(ii) a reduction in the rate and density of M- PA and (iii) no reduction in antibiotic
Beverley Cherie Millar (fig.83 above) is a clinical scientist based at the North of Ireland Public Health Laboratory, Department of Bacteriology, Belfast City Hospital, Belfast UK.and the School of Biomedical Sciences, Ulster University, Coleraine, UK.
Milliken EJT. Cystic Fibrosis in Art. JAMA. 2018 Sep 25;320(12):1224-1226. doi: 10.100he 1/jama.2018.12769. No abstract available.[Pubmed]
-

Fig. 89 Eliza J T Milliken
- This interesting thought-provoking article by Eliza Jane Temple Milliken begins –
“With cystic fibrosis (CF) patients now living well into adulthood, contemporary adult artists with the disease are creating diverse kinds of art, including rap, drawing, performance, video, and sculpture, to express their experiences. The work has characteristic themes of respiratory support and childhood; visual metaphors exploring patient-clinician power dynamics; and it often conveys messages of disempowerment, depersonalisation, and infantilism. Understanding this emerging body of CF art may give physicians insight into CF patients’ experience and the way health care workers and health systems shape it”.
The full text contains examples of art by people with cystic fibrosis (figures below). For readers without full access facilities, examples of the art are reproduced in “Images for Jane Eliza Jane Temple Milliken” on the general internet.
Dr. Eliza Jane Temple Milliken (fig.89) Department of Infectious Diseases and Microbiology, St Vincent’s Hospital, Sydney,
The two pictures on the left are by Grant Gronewald (sunsetgradient@gmail.com). The two pictures on the right are by Dylan Mortimer (www.dylanmortimer.com). All images are reproduced with permission of the artist.

- Never grateful Market research
Montgomery ST, Dittrich AS, Garratt LW, Turkovic L, Frey DL, Stick SM, Mall MA, Kicic A; AREST CF. Interleukin-1 is associated with inflammation and structural lung disease in young children with cystic fibrosis. J Cyst Fibros. 2018 Jun 5. pii: S1569-1993(18)30588-5. doi: 10.1016/j.jcf.2018.05.006. [Epub ahead of print] [Pubmed]

Fig.90 Samuel T Montgomery uwa.edu.au
Bronchoalveolar lavage fluid (BALf) from 102 children with CF were used to determine IL-1α, IL-1β, IL-8 levels and neutrophil elastase (NE) activity, which were then correlated to structural lung changes observed on chest computed tomography (CT) scans. IL-1α and IL-1β were detectable in BAL in absence of infection, increased in the presence of bacterial infection and correlated with IL-8 (p < 0.0001), neutrophils (p < 0.0001) and NE activity (p < 0.01 and p < 0.001). IL-1α had the strongest association with structural lung disease (p < 0.01) in the absence of infection (uninfected: p < 0.01 vs. infected: p = 0.122).
The data associates IL-1α with early structural lung damage in CF and suggests this pathway as a novel anti-inflammatory target.
Samuel T Montgomery (fig.90) is an Honorary Research Associate at the School of Paediatrics and Child Health, University of Western Australia, Nedlands 6009, Western Australia, Australia.
Muhlebach MS, Zorn BT, Esther CR, Hatch JE, Murray CP, Turkovic L, Ranganathan SC, Boucher RC, Stick SM, Wolfgang MC. Initial acquisition and succession of the cystic fibrosis lung microbiome is associated with disease progression in infants and preschool children. PLoS Pathog. 2018 Jan 18;14(1):e1006798. doi: 10.1371/journal.ppat.1006798. eCollection 2018 Jan. Free article available [Pubmed]

Fig. 91 Marianne Muhlebach
eventscribe.net
The authors consider their findings suggest that changes within the CF lower airways microbiome occur during the first years of life and that distinct microbial signatures are associated with the progression of early CF lung disease.
— All children in this study were prescribed amoxicillin-clavulanic acid during the first two years of life, which presumably had some effect on the results, or delayed the onset of lower airways acquisition of oral microbes. Reading of the original paper confirms the importance but great complexity of the subject.
Marianne Muhlebach is a paediatric pulmonologist at the Department of Pediatrics, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina, United States of America.
Mosconi P, Colombo C, Roberto A, Candiani G, Greco MT, Satolli R, Castellani C. Deciding on cystic fibrosis carrier screening: three citizens’ juries and an online survey. Eur J Public Health. 2018 Mar 19. doi: 10.1093/eurpub/cky032. [Epub ahead of print] [Pubmed]

Fig.92 Paola Mosconi
Testing Treatments
A citizens’ jury is one way to ask citizens to deliberate on controversial topics in the interests of a society. The aims of this project were to gather opinions about CF carrier screening through citizens’ jury deliberations and to match them with the findings of a large online consultation survey open to the general population, people with CF and families and health professionals.
Three citizens’ juries and an online survey were asked: ‘Should the Health Service organize screening of the population with the aim of identifying healthy people who may have children with CF?’ The jurors had no medical background and no personal or family CF history. The survey was open to people with CF, families, and healthcare professionals.
Jurors and survey respondents were in favour of CF carrier screening, mainly considering the severity of CF, the value of informed reproductive choices and the equality of the screening. All the citizens’ juries felt positively about the health service actively offer CF carrier screening to provide women and couples of reproductive age equal access and standardized information on the pros and cons.
Considering the favourable attitude towards CF screening, the feasibility of CF screening, in terms of best setting, target age and healthcare professionals providing it, should be tested in a clinical trial.
Paola Mosconi (fig. 92) is Laboratory Head at the Laboratory for Medical Research and Consumers Involvement, Department of Public Health, IRCCS Istituto di Ricerche Farmacologiche Mario Negri, Milan, Italy.
Munck A, Boulkedid R, Weiss L, Foucaud P, Wizla-Derambure N, Reix P, Bremont F, Derelle J, Schroedt J, Alberti C; Gastroenterology and Nutrition Société française de la Mucoviscidose (SFM) Working Group and the ALIMUDE Study Group. Nutritional Status the First Two Years of Life in Cystic Fibrosis Diagnosed by Newborn Screening. J Pediatr Gastroenterol Nutr. 2018 Mar 14. doi: 10.1097/MPG.0000000000001956. [Epub ahead of print] [Pubmed]
-

Fig. 93 Anne Munck
A prospective longitudinal multi-centre study assessing nutritional status according to pancreatic status, feeding modalities, prescriptions, pulmonary outcome and biological nutritional parameters.
One-hundred-and-five infants were recruited and 99 completed the study. Nutritional care management prevented under nutrition and stunting in those with exocrine pancreatic sufficiency (EPS), but affected (13/87) 15% and (21/86) 24%, respectively, of infants with exocrine pancreatic insufficiency (EPI). The logistic regression model found a positive association between both weight and length z-scores “at risk” at month 24, and initial pulmonary symptoms (OR 0.06, p < 0.01 and OR 0.08, p < 0.01, respectively); these symptoms were less frequent when age at first visit was earlier than 1.2 months (33% versus 67%, p = 0.02); stunting was also associated with high-calorie density intake and Staphylococcus aureus (OR: 0.05, p = 0.01 and OR: 0.17, p < 0.01). Pulmonary outcome did not differ according to pancreatic status; breast-feeding for at least 3 months delayed first acquisition of Pseudomonas aeruginosa. Despite sodium and fat-soluble vitamin supplementation, half of both cohorts had low urinary sodium output and half of the EPI cohort had low vitamin D levels.
The authors’ data shed light on the fact that stunting was more frequent than undernutrition, while both parameters involved only patients with pancreatic insufficiency. Modalities of feeding were not associated with nutritional status; breast-feeding may provide some protection against acquisition of Pseudomonas aeruginosa.
– Presumably the significant proportion of infants who are stunted is a manifestation of the intrinsic growth problem reported in a number of previous publications.
Anne Munck (Fig. 93) is a paediatrician at the Service des maladies digestives et respiratoires de l’enfant, CRCM, Hôpital Robert Debré, AP-HP, University Paris Diderot.
Newsome SJ, Daniel RM, Carr SB, Bilton D, Keogh RH.Investigating the effects of long-term dornase alfa use on lung function using registry data. J Cyst Fibros. 2018 Aug 29. pii: S1569-1993(18)30747-1. doi: 10.1016/j.jcf.2018.08.004. [Epub ahead of print]30172681 Full text available. [Pubmed]

Fig. 94 Simon Newsome
Dornase alfa (DNase) is one of the commonest cystic fibrosis treatments and is often used for many years. However, studies have not evaluated the effectiveness of its long-term use. The authors aimed to use UK CF Registry data to investigate the effects of one-, two-, three-, four- and five-years of DNase use on lung function to see if the benefits of short-term treatment use are sustained long term.
They analysed data from 4,198 people in the UK CF Registry from 2007 to 2015 using g-estimation. By controlling for time-dependent confounding they estimated the effects of long-term DNase use on percent predicted FEV1 (ppFEV1) and investigated whether the effect differed by ppFEV1
Considering the population as a whole, there was no significant effect of one-year’s use of DNase; change in ppFEV1 over one year was -0.1% in the treated compared to the untreated (p = 0.51) and this did not change with long-term use. However, treatment was estimated to be more beneficial in people with lower lung function (p < 0.001); those with ppFEV1 < 70% at treatment initiation, showed an increase in lung function over one year that was sustained out to five years. The estimated effect of DNase did not depend on age (p = 0.35). They concluded DNase improved lung function in individuals with reduced lung function, bringing a step-change in lung function, but no change in the slope of decline. There was no evidence for a benefit in lung function in those initiating treatment with ppFEV1 > 70%.
Simon J Newsome (Fig. 94) is a Research Degree Student in Medical Statistics at the London School of Hygiene and Tropical Medicine. He has published previously on cardiovascular topics and the UK CF Registry.
Nap-van der Vlist MM, Burghard M, Hulzebos HJ, Doeleman WR, Heijerman HGM, van der Ent CK, Nijhof SL.Prevalence of severe fatigue among adults with cystic fibrosis: A single center study. J Cyst Fibros. 2018 May;17(3):368-374. doi: 10.1016/j.jcf.2018.03.003. Epub 2018 Mar 29. [Pubmed]
-

Fig. 95 Merel Nap-van der Vlist
- With life expectancy increasing among patients with cystic fibrosis (CF), the prevalence of complications such as fatigue is also expected to increase. Our aim was to investigate the prevalence of severe fatigue among adults with CF and to identify factors associated with fatigue. Adult patients with CF receiving treatment at a single center were invited to complete three questionnaires. We then studied the associations between fatigue and clinically measured parameters and between fatigue and patient-reported outcomes.A total of 77 patients (age 19-54years; 56% males; mean FEV1: 63%) completed the questionnaires (43% response rate). The prevalence of severe fatigue among these patients was 26%. The variance in fatigue was explained partially by clinically measured parameters. However, patient-reported outcomes were stronger independently associated with fatigue and included the patients’ reported respiratory symptoms, emotional functioning, and social functioning. Fatigue is a clinically important and highly prevalent issue among adults with CF and is associated with a significant reduction in health-related quality of life and participation in society. In addition, fatigue is associated more strongly with the patient’s perception of symptoms and well-being than with clinically measured parameters.
Dr Merel M Nap-van der Vlist (fig. 95) is a medical doctor and PhD student in the Department of Pediatrics, Wilhelmina Children’s Hospital, University Medical Center, Utrecht
Letter relating to the above article of Nap-van der Vlist MM et al.
Talbot NP, Flight WG. Anaemia and iron deficiency in relation to fatigue in cystic fibrosis. Cyst Fibros. 2019 Jan;18(1):e5. doi: 10.1016/j.jcf.2018.08.002. Epub 2018 Sep 6. Letter [Pubmed]

Fig.96 Nick Talbot dpag.ox.ac.uk
The authors welcomed Nap-van der Vlist and colleagues’ recent paper on severe fatigue in people with cystic fibrosis. However, they suggest the authors have not accounted for common and potentially treatable causes of fatigue in CF, namely anaemia and iron deficiency. Iron deficiency has been shown repeatedly to be very common in CF, affecting up to 74% of adults, with anaemia seen in as many as 29%. Although Jarad and colleagues found no statistical correlation between fatigue and haemoglobin levels, their data were drawn from a small, single-centre cohort of only 44 patients and no information on iron status was provided.
Identification and correction of iron deficiency in CF is a challenge. Gifford and colleagues found no benefit in haemoglobin levels following six weeks of oral iron supplementation in adults with CF. Hoo and Wildman have previously raised concerns over the safety of intravenous iron therapy in CF having reported a high rate of clinical deterioration following IV iron infusion in a small case series of five patients. Conversely, however, correction of iron deficiency using intravenous iron has been shown to improve fatigue and exercise tolerance in a variety of other chronic conditions such as heart failure and rheumatoid arthritis, even in the absence of anaemia, with no safety signal seen in randomised controlled trials of IV iron in the setting of critical illness.
— These authors consider it would be of great interest to interpret the rates of fatigue seen in the study by Nap-van der Vlist and colleagues with the addition of data on iron deficiency and anaemia. They suggest there is a clear need for prospective studies of intravenous iron therapy in people with CF, which has the potential to have a significant impact on the wellbeing and fatigue levels in this group.
Dr Nick Talbot is a Non‐Stipendiary Lecturer in Biomedical Sciences and Research Fellow, St Peter’s College Oxford and also the Nuffield Department of Medicine, University of Oxford, United Kingdom; Oxford University Hospitals NHS Foundation Trust, United Kingdom.
Dr W G Flight is Director of the Adult CF Unit , Oxford University Hospitals NHS Foundation Trust, United Kingdom
Stephanie C M Nijmeijer, Thirsa Conijn, Phillis Lakeman, Lidewij Henneman, Frits A Wijburg, Lotte Haverman. Attitudes of the general population towards preconception expanded carrier screening for autosomal recessive disorders including inborn errors of metabolism. Mol Genet Metab . 2019 Jan;126(1):14-22.doi: 10.1016/j.ymgme.2018.12.004. Epub 2018 Dec 10.[Pubmed]

Fig. 97 Stephanie Nijmeijer ResearchGate
Background: A substantial number of severely debilitating and often ultimately fatal inborn errors of metabolism (IEMs) still lack an effective disease-modifying treatment. Informing couples before a pregnancy about an increased risk of having a child with an inherited disorder is now feasible by preconception expanded carrier screening (ECS). While knowledge about carrier status enhances reproductive autonomy, it may also result in ethical dilemmas.
The purpose of this study was to assess the attitudes of the general Dutch population towards preconception ECS and to investigate which factors influence these attitudes.
Methods: Data collection was carried out in collaboration with a market research agency. In total, 1188 Dutch individuals of reproductive age (18-45 years) were invited by email to complete an online ECS questionnaire in 2016. Prior to the start of the questionnaire, a written explanation of the concepts of autosomal recessive (AR) inheritance, carrier status and ECS was presented.
Results: The questionnaire was completed by 781 individuals (65.7%), of whom 31% indicated they would take an ECS test themselves. In addition, 55% agreed that ECS should be offered to all prospective parents. The most frequently selected argument in favor of ECS (47.2%) was that participants want to spare a child from a life with a severe hereditary disorder. The reason most often mentioned not to participate in ECS (48%) was that participants reported not having a hereditary disorder in the family. The majority preferred receiving individual test results above a couple-based disclosure method in which participants receive the carrier status results only when they are a carrier couple of the same disorder. Participants with religious beliefs were less likely to participate in ECS, whereas participants who were considering a (future) pregnancy were more likely to participate.
Conclusion: Our study demonstrates an overall positive attitude among participants of reproductive age in the general Dutch population towards preconception ECS. A striking misconception is that many of the participants believe that ECS is of interest only for those with a positive family history of one of the hereditary disorders. This finding emphasizes the importance of providing understandable, balanced information and education to the general public regarding the concepts of inheritance when presenting the option of carrier screening. Our results provide valuable insights that can be used in the debate about the responsible implementation of preconception expanded carrier screening or autosomal recessive disorders, including inborn errors of metabolism.
— These results are not unexpected as there is only a modest interest on the part of the general public in preconception and antenatal screening.
Dr Stephanie C M Nijmeijer (fig.9 7) Amsterdam UMC, University of Amsterdam, Pediatric Metabolic Diseases, Emma Children’s Hospital and Amsterdam Lysosome Center “Sphinx”, Amsterdam, Netherlands.
O’Connor K, Jukes T, Goobie S, DiRaimo J, Moran G, Potter BK, Chakraborty P, Rupar CA, Gannavarapu S, Prasad C .Psychosocial impact on mothers receiving expanded newborn screening results. Eur J Hum Genet. 2018 Apr;26(4):477-484. doi: 10.1038/s41431-017-0069-z. Epub 2018 Jan 29. [Pubmed]
Expanded newborn screening (NBS) for genetic disorders has improved diagnosis of numerous treatable diseases, positively impacting children’s health outcomes. However, research about the psychological impact of expanded NBS on families, especially mothers, has been mixed.
Our study examined associations between maternal experiences of expanded NBS and subsequent psychosocial functioning and parenting stress in mothers whose infants received either true negative (TN), true positive (TP) or false positive (FP) results after a 4- to 6-month period. The Parenting Stress Index and the Depression, Anxiety and Stress Scale were used to assess symptoms of anxiety, stress and depression in 3 sets of mothers, whose infants received TN (n = 31), TP (n = 8) or FP (n = 18) results. Multivariate analyses of variance (MANOVA) results revealed no significant differences among these three groups of mothers regarding overall anxiety, stress and depression. However, FP mothers experienced lower levels of stress related to their own health compared to TN group. Two potential trends were also identified; results suggested TN mothers might experience higher levels of isolation than mothers in the TP group and that FP mothers might report higher stress levels in relation to spousal relationships compared to the TN group. FP mothers seemed to report similar or better levels of psychosocial functioning than TN mothers.
The authors consider their findings are encouraging with respect to impacts of NBS on maternal well-being. They also identify key areas for improvement (parental education) and research (isolation and spousal relationships).
Kathleen A O’Connor is at the Department of Psychology Western University London, Ontario.
Pagaduan JV, Ali M, Dowlin M, Suo L, Ward T, Ruiz F, Devaraj S. Revisiting sweat chloride test results based on recent guidelines for diagnosis of cystic fibrosis. Pract Lab Med. 2018 Jan 3;10:34-37. doi: 10.1016/j.plabm.2018.01.001. eCollection 2018 Mar[Pubmed]

Fig. 98 Jayon Pagaduan
Recent sweat chloride guidelines published by the Cystic Fibrosis Foundation changed the intermediate sweat chloride concentration range from 40-59 mmol/L to 30-59 mmol/L for age > 6 months. The authors wished to determine how this new guideline would impact detection of cystic fibrosis among patients who previously had sweat tests done at Texas Children’s Hospital. They revisited sweat chloride test results (n = 3012) in the last 5 years at Texas Children’s Hospital based on the new guidelines on diagnosis of cystic fibrosis from the Cystic Fibrosis Foundation.
They identified 125 patients that would be reclassified in the intermediate sweat chloride value with the new guidelines that were classified as “unlikely to have CF” in the previous guidelines. 8 (32%) patients with CFTR gene testing were positive for CFTR gene mutation(s). 4 (50%) of these patients were identified to have 2 CFTR mutations. One had variant combination that was reported to cause CF but all were diagnosed with CFTR-related metabolic syndrom
Their findings concur with the new CF diagnosis guidelines that changing the intermediate cut-off to 30-59 mmol/L sweat chloride concentration in combination with CFTR genetic analysis enhances the probability of identifying individuals that have risk of developing CF or have CF and enables for earlier therapeutic intervention.
Jayson V Pagaduan (fig. 98) is a chemical pathologist in the Department of Pathology & Immunology, Baylor College of Medicine, Department of Pathology and Immunology, Texas Children’s Hospital, Houston, TX 77030, USA .
Pallin M, Keating D, Kaye DM, Kotsimbos T, Wilson JW.Subclinical Left Ventricular Dysfunction is Influenced by Genotype Severity in Patients with Cystic Fibrosis.Clin Med Insights Circ Respir Pulm Med. 2018 Aug 19;12:1179548418794154. doi: 10.1177/1179548418794154. eCollection 2018. pubmed.ncbi.nlm.nih.gov/30147387/
Over 2000 genotypes in the cystic fibrosis (CF) gene have been described. These genotypic differences result in variable clinical manifestations of CF, with severity of disease dependent on CF transmembrane conductance (CFTR) protein function. CFTR is widely distributed in nucleated cells, including cardiac myocytes, but the effect of genotype on cardiac function. This retrospective review of echocardiographic data is from a single adult CF centre between 2000 and 2015. Patients were cohorted based on the functional classification of genotype. ‘Severe’ patients had both CF genes from functional classification groups 1-3; ‘mild’ patients had one or no gene from these groups, or in the event of the second gene being unknown were pancreatic sufficient. Genotype and echocardiography were recorded during the inclusion period in 100 patients, 79 of whom were classified as having severe genotypes. Although the severe group were younger they had a lower fractional shortening (33.66 ± 6.6 vs 36.9 ± 6.3, P < .05), left atrial area (14.9 ± 3.6 versus 18.0 ± 4.2 cm2; P < .01) and volume (39.9 ± 18.7 versus 51.0 ± 18.7 mL; P < .05) and showed a trend to lower left ventricular ejection fraction.
This study is the first to show that in CF, severity of genotype (functional classification) is associated with cardiac impairment. Patients with severe CF genotype and cardiac dysfunction should be identified to evaluate cardiac response to gene-modifying treatments prior to consideration for lung transplantation.
Michael Pallin is with the Cystic Fibrosis Service, Alfred Health and Central Clinical School, Monash University, Melbourne, VIC, Australia.
Pica F, Gaziano R, Casalinuovo IA, Moroni G, Buè C, Limongi D, D’Agostini C, Tomino C, Perricone R, Palamara AT, Sinibaldi Vallebona P, Garaci E.Serum thymosin alpha 1 levels in normal and pathological conditions.Expert Opin Biol Ther. 2018 Jul;18(sup1):13-21. doi: 10.1080/14712598.2018. [Pubmed]

Fig. 99 Francesca Pica
UniCamillus
Thymosin alpha 1 (Ta1) is a natural occurring peptide hormone that is crucial for the maintenance of the organism homeostasis. It has been chemically synthesized and used in diseases where the immune system is hindered or malfunctioning. Areas covered: Many clinical trials investigate the Ta1 effects in patients with cancer, infectious diseases and as a vaccine enhancer. The number of diseases that could benefit from Ta1 treatment is increasing. To date, questions remain about the physiological basal levels of Ta1 and the most effective dose and schedule of treatment. Evidence is growing that diseases characterized by deregulation of immune and/or inflammatory responses are associated with serum levels of Ta1 significantly lower than those of healthy individuals: to date, B hepatitis, psoriatic arthritis, multiple sclerosis and sepsis. The sputum of cystic fibrosis patients contains lower levels of Ta1 than healthy controls. These data are consistent with the role of Ta1 as a regulator of immunity, tolerance and inflammation. Expert opinion: Low serum Ta1 levels are predictive and/or associated with different pathological conditions. In case of Ta1 treatment, it is crucial to know the patient’s baseline serum Ta1 level to establish effective treatment protocols and monitor their effectiveness over time.
Dr. Francesca Pica (fig.99) is Associate Professor of Microbiology and Clinical Microbiology, Departments of Experimental Medicine and Surgery, University of Rome Tor Vergata, Rome, Italy.
Also see Garaci E. From thymus to cystic fibrosis: the amazing life of thymosin alpha 1. Expert Opin Biol Ther. 2018 Jul;18(sup1):9-11. doi: 10.1080/14712598.2018.1484447.[Pubmed]
Préville-Ratelle S, Coriati A, Ménard A, Bourdeau I, Tremblay F, Berthiaume Y. Adrenal Insufficiency in Cystic Fibrosis: A Rare Phenomenon? Can Respir J. 2018 Mar 13;2018:3629031. doi: 10.1155/2018/3629031. eCollection 2018. [Pubmed]

Fig. 100 Yves Berthiaume Research Gate
The prevalence of adrenal insufficiency (AI) in cystic fibrosis (CF) is unknown. The frequent use of glucocorticoids (inhaled or systemic) may induce the long-term suppression of the hypothalamic-pituitary-adrenal axis. The authors reviewed the results of adrenocorticotropic hormone (ACTH) stimulation tests done over a 10-year period to evaluate adrenal function in 69 CF patients of the CHUM CF clinic. Clinical characteristics of AI patients were compared to adrenal-sufficient (AS) patients.
Adrenal insufficiency was confirmed in 33 of the 69 CF patients. A higher rate of dysglycemia (P=0.022) and of Aspergillus positive culture (P=0.006) was observed in AI patients compared to AS patients. Weight, CFTR genotype, and pulmonary function were comparable between AI and AS patients. The use of systemic corticosteroids (SC) prior to the diagnosis of AI was observed in 42.4% of patients, but none of the AS patients. Inhaled corticosteroids in 67% of the AS and 94% of the AI patients. Compared to AI patients without SC, SC-treated AI patients were older and had a higher rate of allergic bronchopulmonary aspergillosis
This study is the first to systematically examine the presence of AI in the largest cohort of CF patients studied to date with a prevalence of 8%. Patients treated with corticosteroids and those colonized with Aspergillus have a greater risk of AI.
Corresponding author is Professor Yves Berthiaume (Fig. 100) Institut de recherches cliniques de Montréal (IRCM),and Clinique de fibrose kystique, Centre hospitalier de l’Université de Montréal (CHUM), Canada H2X.
Quon BS, Sykes J, Stanojevic S, Marshall BC, Petren K, Ostrenga J, Fink A, Elbert A, Faro A, Goss CH, Stephenson AL.Clinical characteristics of cystic fibrosis patients prior to lung transplantation: An international comparison between Canada and the United States.Clin Transplant. 2018 Jan 2. doi: 10.1111/ctr.13188. [Epub ahead of print [Pubmed]
-

Fig. 101 Bradley S Quon
- Between 1986 and 2013, 607 (10.2%) CF patients underwent lung transplantation in Canada and 3428 (7.5%) in the United States. A lower proportion of recipients had growth of B. cepacia complex prior to transplant in the United States compared to Canada (0.8% vs 4.3%). Lung function was similar between recipients from the two countries. The proportion of patients classified as underweight was significantly higher in the United States compared to Canada (39.8% vs 28.0%; SD 26.1) despite higher rates of feeding tube use (42.5% vs 28.6%; SD 29.0).
CF lung transplant recipients from the United States have similar lung function, lower rates of B. cepacia complex, and worse nutritional parameters prior to transplant compared to counterparts in Canada. Future studies are necessary to evaluate the impact of these differences on post-transplant survival.
Dr. Bradley S Quon (fig.101 is Assistant Professor in the Department of Medicine University of British Columbia. He is a Staff Respirologist and CF Physician at St. Paul’s Hospital in Vancouver
– Presumably the better nutrition and higher B. cepacia rates are a reflection of the patients’ previous experience – the Canadians being better nourished for decades from the time of Douglas Crozier in the Seventies but more likely to have B. cepacia which became a common pathogen in Toronto patients from the Eighties
Ratjen F, Moeller A, McKinney ML, Asherova I, Alon N, Maykut R, Angyalosi G; EARLY study group. Collaborators (12)Eradication of early P. aeruginosa infection in children <7 years of age with cystic fibrosis: The early study. J Cyst Fibros. 2018 Apr 20. pii: S1569-1993(18)30087-0. doi: 10.1016/j.jcf.2018.04.002. [Epub ahead of print] [Pubmed]

Fig. 102 Felix Ratjen
Cochrane Cystic Fibrosis
Antibiotic eradication treatment is the standard-of-care for cystic fibrosis (CF) patients with early Pseudomonas aeruginosa (Pa)-infection; however, evidence from placebo-controlled trials is limited. This double-blind, placebo-controlled trial randomised CF patients aged 3 months to <7 years (N = 51) with early Pa-infection to tobramycin inhalation solution (TOBI 300 mg) or placebo (twice daily) for 28 days with an optional cross-over on Day 35. Primary endpoint was proportion of patients having throat swabs/sputum free of Pa on Day 29.
On Day 29, 84.6% patients in the TOBI versus 24.0% in the placebo group were Pa-free (p < 0.001). At the end of the cross-over period, 76.0% patients receiving TOBI in the initial 28 days were Pa-free compared to 47.8% receiving placebo initially. Adverse events were consistent with the TOBI safety profile with no differences between TOBI and placebo.
The authors concluded TOBI was effective in eradicating early Pa-infection with a favourable safety profile in young CF patients.
– Not mentioned in the summary was the fact that at day 91 more patients receiving TOBI during the initial 28 days (the TOBI/placebo group:19/25 patients, 76%) were Pa free compared with those initially on placebo (placebo/TOBI group: 11/23 patients 47.8 %). This was unfortunate for the latter group and emphasises the point that eradication treatment must be early – the greater the delay in initiating treatment the more the risk of chronic Pa infection as occurred in these placebo-treated children. These facts were already known however apparently this trial has satisfied the regulatory requirements.
Felix Ratjen (fig. 102) is professor in the Division of Respiratory Medicine, Department of Pediatrics, Translational Medicine, Hospital for Sick Children, University of Toronto, Toronto, ON, Canada.
Ren CL, Morgan RL, Oermann C, Resnick HE, Brady C, Campbell A, DeNagel R, Guill M, Hoag J, Lipton A, Newton T, Peters S, Willey-Courand DB, Naureckas ET.Cystic Fibrosis Foundation Pulmonary Guidelines: Use of CFTR Modulator Therapy in Patients with Cystic Fibrosis. Ann Am Thorac Soc. 2018 Jan 17. doi: 10.1513/AnnalsATS.201707-539OT. [Epub ahead of print[Pubmed]

Fig 103 Clement L Ren CHOP Research Institute
A multidisciplinary committee of CF caregivers and patient representatives was assembled. A systematic review was conducted to find relevant publications. The systematic review yielded 6 publications applicable to our clinical questions.
In summary their recommendations were as follows – “For adults and children aged 6 years and older with CF due to gating mutations other than G551D or R117H,(i.e G178R, S549N, S549R, G551S, G1244E, S1251N, S1255P, or G1349D)the guideline made a conditional recommendation for treatment with ivacaftor (IVA) for 1) adult aged 18 years and older and 2) for children aged 6-17 years with an FEV1 of less than 90% predicted. For those with the R117H mutation, the guideline made a conditional recommendation against treatment with IVA for 1) children aged 2-17 years with an FEV1 greater than 90% predicted and 2) children less than 6 years of age. Among those with two copies of F508del the guideline panel made a strong recommendation for treatment with ivacaftor/lumacaftor for 1) adults and children 12 years and older with an FEV1 greater than 90% predicted and 2) children aged 6-11 years”.
The authors suggest that these recommendations will be of help to CF clinicians, patients, and their families in guiding decisions regarding use of these medications.
Clement L Ren (fig 103) at the time at Indiana University School of Medicine, Indianapolis, Indiana.
Rodriguez Miguelez P, Seigler N, Tucker MA, Csanyi G, McKie KT, Forseen C, Harris RA. Sildenafil improves vascular endothelial function in patients with cystic fibrosis. Am J Physiol Heart Circ Physiol. 2018 Aug 31. doi: 10.1152/ajpheart.00301.2018. [Epub ahead of print] [Pubmed]
-

Fig. 104 Paula Rodriquez-Miguelez Department of Kinesiology
- Cystic fibrosis (CF), characterized by defective CFTR function, is associated with multiple systemic complications including vascular dysfunction. Sildenafil, a phosphodiesterase type 5 (PDE5) inhibitor, not only enhances nitric oxide (NO) metabolism, it has been shown to improve CFTR functionality as well. Thus, sildenafil has been proposed as a potential therapy to improve vascular health in CF; however, its potential therapeutic role has yet to be determined.
To investigate the effect of sildenafil treatment on endothelial function in patients with CF.
Patients with CF completed a randomized, double blind, placebo controlled, crossover study with an acute dose of sildenafil (50 mg) and placebo followed by a four-week, open label extension with sildenafil (20 mg/day). Flow-mediated dilation (FMD) was used to evaluate endothelial function before and after treatments. In addition, phosphorylated (p) and total endothelial NO synthase (NOS3) protein expression was determined from endothelial cells that were exposed to plasma from the patients before and after four weeks of sildenafil.
No changes (p≥0.110) in endothelial function were observed after the acute dose of sildenafil. However, FMD significantly (p=0.029) increased after four weeks of treatment (∆FMD= 1.5 ± 2.2%). Moreover, p-NOS3 protein expression significantly (p=0.013) increased following 4 weeks of treatment (∆p-NOS3= 0.31 ± 0.39 AU) and was associated (r=0.593; p=0.033) with the observed change in FMD.
These data suggest that four weeks of sildenafil treatment can improve vascular endothelial function in patients with CF, likely through an increase in NOS3 phosphorylation.
Paula Rodriguez-Miguelez (fig.104) is Assistant Professor of Health Science, Department of Kinesiology and Health Sciences at Virginia Commonwealth University, Richmond
Romani L, Oikonomou V, et al. Publisher Correction: Thymosin α1 represents a potential potent single-molecule-based therapy for cystic fibrosis. Nat Med. 2018 Sep;24(9):1482. doi: 10.1038/s41591-018-0099-2.
In the version of this article originally published, the amino acid sequence for Tα1 described in the Online Methods is incorrect. The sequence is described as “Ac-SDAAVDTSSEITTJDLKEKKEVVEEAEN-OH”. It should be “Ac-SDAAVDTSSEITTKDLKEKKEVVEEAEN-OH”. The error has been corrected in the HTML and PDF versions of this article.
Ronan NJ, Einarsson GG, Twomey M, Mooney D, Mullane D, NiChroinin M, O’Callaghan G, Shanahan F, Murphy DM, O’Connor OJ, Shortt CA, Tunney MM, Eustace JA, Maher MM, Elborn JS, Plant BJ. CORK Study in Cystic Fibrosis: Sustained Improvements in Ultra-Low-Dose Chest CT Scores After CFTR Modulation With Ivacaftor. Chest. 2018 Feb;153(2):395-403.doi: 10.1016/j.chest.2017.10.005. Epub 2017 Oct 14[Pubmed]

Fig. 105 Nicola J Ronan. Irish Thoracic Society
Ivacaftor produces significant clinical benefit in patients with cystic fibrosis (CF) with the G551D mutation. Prevalence of this mutation at the Cork CF Centre is 23%. This study assessed the impact of cystic fibrosis transmembrane conductance regulator modulation on multiple modalities of patient assessment. Thirty-three patients with the G551D mutation were assessed at baseline and prospectively every 3 months for 1 year after initiation of ivacaftor. Change in ultra-low-dose chest CT scans, blood inflammatory mediators, and the sputum microbiome were assessed. Significant improvements in FEV1, BMI, and sweat chloride levels were observed post-ivacaftor treatment. Improvement in ultra-low-dose CT imaging scores were observed after treatment, with significant mean reductions in total Bhalla score (P < .01), peribronchial thickening (P = .035), and extent of mucous plugging (P < .001). Reductions in circulating inflammatory markers, including interleukin (IL)-1β, IL-6, and IL-8 were demonstrated. There was a 30% reduction in the relative abundance of Pseudomonas species and an increase in the relative abundance of bacteria associated with more stable community structures. Post treatment community richness increased significantly (P = .03).
The authors concluded early and sustained improvements on ultra-low-dose CT scores suggest it may be a useful method of evaluating treatment response. It paralleled improvement in symptoms, circulating inflammatory markers, and changes in the lung microbiota.
Nicola J Ronan (Fig.105 ) is at the Cork Cystic Fibrosis Centre, Cork University Hospital, University College Cork, Cork, Ireland; HRB Clinical Research Facility, Cork University Hospital, University College Cork, Cork, Ireland.
Ronchetti K, Tame JD, Paisey C, Thia LP, Doull I, Howe R, Mahenthiralingam E. The CF-Sputum Induction Trial (CF-SpIT) to assess lower airway bacterial sampling in young children with cystic fibrosis: a prospective internally controlled interventional trial.Lancet Respir Med.2018 Jun;6(6):461-471. doi: 10.1016/S2213-2600(18)30171-1. Epub 2018 May 16. pubmed.ncbi.nlm.nih.gov/29778403/

Fig.107 JoDee Tame
@Jodeetame

Fig.106 Katherine Rochetti
healthcharity.wales
A large study from the Paediatric Unit in Cardiff to assess the pathogen yield from sputum induction compared with that from cough swab and single- lobe, two-lobe, and six-lobe bronchoalveolar lavage in children with cystic fibrosis aged between 6 months and 18 years. Samples from cough swab, sputum induction, and single-lobe, two-lobe, and six-lobe bronchoalveolar lavage were matched for within-patient comparisons. Primary outcomes were comparative pathogen yield between sputum induction and cough swab for stage 1, and between sputum induction, and single-lobe, two-lobe, and six-lobe bronchoalveolar lavage for stage 2.
Between Jan 23, 2012, and July 4, 2017, 124 patients were prospectively recruited to the trial and had 200 sputum induction procedures for stage 1. 167 (84%) procedures were successful and the procedure was well tolerated.
Of the 167 paired samples, 63 (38%) sputum-induction samples were pathogen positive compared with 24 (14%) cough swabs (p<0·0001; odds ratio [OR] 7·5; 95% CI 3·19-17·98). More pathogens were isolated from sputum induction than cough swab (79 [92%] of 86 vs 27 [31%] of 86; p<0·0001).
For stage 2, 35 patients had a total of 41 paired sputum-induction and bronchoalveolar lavage procedures. Of the 41 paired samples, 28 (68%) were positive for at least one of the concurrent samples. 39 pathogens were isolated. Sputum induction identified 27 (69%) of the 39 pathogens, compared with 22 (56%; p=0·092; OR 3·3, 95% CI 0·91-12·11) on single-lobe, 28 (72%; p=1·0; OR 1·1, 95% CI 0·41-3·15) on two-lobe, and 33 (85%; p=0·21; OR 2·2, 95% CI 0·76-6·33) on six-lobe bronchoalveolar lavage.
Sputum induction is superior to cough swab for pathogen detection, is effective at sampling the lower airway, and is a credible surrogate for bronchoalveolar lavage in symptomatic children. A substantial number of bronchoscopies could be avoided if sputum induction is done first and pathogens are appropriately treated. Both sputum induction and six-lobe bronchoalveolar lavage provide independent, sizeable gains in pathogen detection compared with the current gold-standard two-lobe bronchoalveolar lavage. The authors propose that sputum induction and six-lobe bronchoalveolar lavage combined are used as standard of care for comprehensive lower airway pathogen detection in children with cystic fibrosis.
Katherine Ronchetti (fig.106) and Jo-Dee Tame (Fig. 107) are physiotherapists at the Children’s Hospital for Wales. Department of Paediatric Respiratory Medicine, and the Department of Paediatric Physiotherapy, Noah’s Ark Children’s Hospital for Wales, Cardiff, UK.
See comments by Andre Schultz below
Rosenfeld M,Wainwright CE,Higgins M,Wang LT,McKee C,Campbell D,Tian S,Schneider J,Cunningham S,Davies JC;ARRIVAL study group.Ivacaftor treatment ofcystic fibrosisin children aged 12 to <24 months and with a CFTR gating mutation (ARRIVAL): a phase 3 single-arm study. Lancet Respir Med.2018 July;^(7):545-553. doi: 10.1016/S2213-2600(18)30202-9. [Epub ahead of print] 2018 Jun 7. [Pubmed]
-

Fig. 108 Margaret Rosenfield
- The ARRIVAL study is a phase 3, single-arm, two-part, multicentre study. Eligible children were aged 12 to <24 months at enrolment and had a confirmed diagnosis of cystic fibrosis and a CFTR gating mutation on at least one allele and could participate in one or both parts of the study.
Children received 50 mg (bodyweight 7 to <14 kg) or 75 mg (bodyweight ≥14 to <25 kg) ivacaftor orally every 12 h. In study part A, children received ivacaftor for 3 days plus one morning. In study part B, children received 24 weeks of treatment. Seven children were enrolled in part A, of whom five received 50 mg and two received 75 mg ivacaftor. All completed treatment. Of 19 children enrolled in part B, including one from part A, all received 50 mg ivacaftor and 18 completed treatment (one withdrew because of difficulty with blood draws). All children received at least one dose of ivacaftor. Pharmacokinetics indicated exposure was similar to that in children aged 2 to <6 years and adults. No children discontinued because of adverse events or safety findings. In part A, three (43%) of seven children had treatment-emergent adverse events, all of which were mild and deemed not to be or unlikely to be related to ivacaftor. By 24 weeks in part B, treatment-emergent adverse events had been reported in 18 (95%) of 19 children, of which most were mild or moderate and the most frequent was cough (14 [74%] children). Two children in part B had four serious adverse events: one had constipation (possibly related to ivacaftor), distal intestinal obstruction syndrome, and eczema herpeticum, and one had persistent cough, all needing hospital admission. In five (28%) of 18 children aspartate or alanine aminotransferase concentrations rose to more than three times the upper limit of normal (to more than eight times in two children with concurrent infections). At week 24, the mean absolute change from baseline in sweat chloride concentration was -73·5 (SD 17·5) mmol/L. Growth parameters for age were normal at baseline and at week 24. At week 24, concentrations of faecal elastase-1 had increased and concentrations of immunoreactive trypsinogen had decreased from baseline. Mean serum lipase and amylase were raised at baseline and rapidly decreased after treatment was started.
The authors concluded that ivacaftor was generally safe and well tolerated in children aged 12 to <24 months for up to 24 weeks and was associated with rapid and sustained reductions in sweat chloride concentrations. Improvements in biomarkers of pancreatic function suggest that ivacaftor preserves exocrine pancreatic function if started early. The study is continuing in infants younger than 12 months.
Margaret Rosenfield (fig. 108) is a paediatrician in the Department of Pediatrics, Seattle Children’s Hospital, University of Washington School of Medicine, Seattle, WA, USA.
Rousset-Jablonski C, Reynaud Q, Nove-Josserand R, Ray-Coquard I, Mekki Y, Golfier F, Durieu I; Study conducted in Lyon France. High proportion of abnormal pap smear tests and cervical dysplasia in women with cystic fibrosis. Eur J Obstet Gynecol Reprod Biol. 2018 Feb;221:40-45. doi: 10.1016/j.ejogrb.2017.12.005. Epub 2017 Dec 9 [Pubmed]

Fig. 109 Christine Rousset Jablonski
Insufficient gynaecological follow-up and cervical screening has been reported in women with cystic fibrosis. Some of these patients will require a pulmonary transplantation, known to be associated with a higher risk of cervical dysplasia. The aim of this study was to explore the results of cervical screening in adult women with CF, and to report the prevalence of abnormal pap smear tests in this population. The authors retrospectively analysed medical records of sexually active women with CF who attended a gynaecological consultation in Lyon University CF referral centre between June 2014 and December 2015. The primary outcome was the result of the pap smear test. There were 176 women attending the CF centre of whom 51 attended for a consultation with the gynaecologist Eventually 47 women (32 non-transplanted and 15 transplanted) were included in the study. The median age of the patients was 28 (range 18-53). The clinical examination revealed that 20 (42.5%) women presented an abnormality (inflammatory cervix, cervical or vulvo-vaginal condyloma). An abnormal pap smear was found in 8/32 (25%) non transplanted women and in 5/15 (33.3%) transplanted women, with no significant difference between the two groups (p=0.75): seven atypical squamous cells of undetermined significance (ASC-US), five low grade squamous intraepithelial lesion (LSIL), one atypical glandular cells (AGC). Six (12.8%) (four non transplanted, and two transplanted) women had an histologically proven dysplasia (four Cervical Intraepithelial Neoplasia (CIN)1, one CIN2, and one endocervical adenocarcinoma in situ). Overall, ten (21.3%) women had a Human Papilloma Virus (HPV) related disease (cervical and/or vulvo-vaginal).
The authors concluded a high proportion of both transplanted and non-transplanted women with CF had abnormal pap smear tests and cervical dysplasia. The strongly recommended a regular gynaecological follow-up, periodic cervical screening, and routine HPV vaccination in this population.
Dr. Christine Rousset-Jablonski (fig 109) of the Centre Léon Bérard, Lyon involved in General Medicine, Oncology, Gynaecology.
Sala MA, Jain M. Tezacaftor for the treatment of cystic fibrosis. Expert Rev Respir Med. 2018 Sep;12(9):725-732. doi: 10.1080/17476348.2018.1507741. Epub 2018 Aug 9. [Pubmed]

Fig 110 Marc Sala
nm.org/doctors
Cystic fibrosis (CF) is the most common, life-limiting autosomal recessive disease in Caucasians, and is caused by defects in production of the CFTR ion channel. Until recently, there were no available treatments targeting the disease-causing defects in CFTR but newly developed CFTR modulators are changing the course of disease in CF. The newest modulator, tezacaftor, is a CFTR corrector that was recently approved by the FDA to be used in combination with the first approved CFTR potentiator, ivacaftor. Areas covered: A detailed review of the clinical trials and published literature, focusing on safety and efficacy, leading to the approval of tezacaftor in CF. Expert commentary: Recent trials have demonstrated that the combination of tezacaftor-ivacaftor is a slightly superior combination to its predecessor, lumacaftor-ivacaftor, with respect to an increase in FEV1, adverse event profile, and drug-drug interactions. It is also approved for a large number of non-F508del, residual function mutations that are predicted to respond based on in vitro testing. The horizon for continued improvements in CFTR-targeted treatments is promising, with three-drug combinations currently in Phase 3 clinical trials, and other drugs with novel mechanisms of action being studied. Within the next 5 years, the vast majority of patients with CF are expected to have a modulator approved for their genotype.
Marc A Sala (fig. 110) is assistant professor in the Division of Pulmonary and Critical Care Medicine, Feinberg School of Medicine , Northwestern University , Chicago , IL , USA.
Southern KW, Patel S, Sinha IP, Nevitt SJ.Correctors (specific therapies for class II CFTR mutations) for cystic fibrosis.Cochrane Database Syst Rev. 2018 Aug 2;8:CD010966. doi: 10.1002/14651858.CD010966.pub2. [Epub ahead of print] [Pubmed]

Fig. 111 Kevin Southern
After a detailed consideration of the available published evidence (please see full abstract for details) the authors concluded there was insufficient evidence that monotherapy with correctors has clinically important effects in people with CF who have two copies of the F508del mutation. Combination therapies (lumacaftor-ivacaftor and tezacaftor-ivacaftor) each result in similarly small improvements in clinical outcomes in people with CF; specifically improvements quality of life (moderate-quality evidence), in respiratory function (high-quality evidence) and lower pulmonary exacerbation rates (moderate-quality evidence). Lumacaftor-ivacaftor is associated with an increase in early transient shortness of breath and longer-term increases in blood pressure (high-quality evidence). These adverse effects were not observed for tezacaftor-ivacaftor. Tezacaftor-ivacaftor has a better safety profile, although data are not available for children younger than 12 years. In this age group, lumacaftor-ivacaftor had an important impact on respiratory function with no apparent immediate safety concerns, but this should be balanced against the increase in blood pressure and shortness of breath seen in longer-term data in adults when considering this combination for use in young people with CF.
Professor Kevin Southern (fig.111) is Professor of Paediatrics at the Alder Hey Children’s Hospital and Liverpool University.
Sanders DB, Zhang Z, Farrell PM, Lai HJ; Wisconsin CF Neonatal Screening Group. Early life growth patterns persist for 12 years and impact pulmonary outcomes in cystic fibrosis.J Cyst Fibrosis 2018 Jan 30. pii: S1569-1993(18)30009-2. doi: 10.1016/j.jcf.2018.01.006. [Epub ahead of print] [Pubmed]

Fig. 112 Don B Sanders Indiana School of medicine
In children with cystic fibrosis (CF), recovery from growth faltering within 2 years of diagnosis (Responders) is associated with better growth and less lung disease at age 6 years. This study examined whether theses benefits are sustained through to the age of 12 years. Longitudinal growth from 76 children with CF enrolled in the Wisconsin CF Neonatal Screening Project was examined and categorised into 5 groups: R12, R6, and R2, representing Responders who maintained growth improvement to age 12, 6, and 2 years, respectively, and I6 and N6, representing Non-responders whose growth did and did not improve during ages 2-6 years, respectively. Lung disease was evaluated by % predicted forced expiratory volume in one second (FEV1) and chest radiograph (CXR) scores. Sixty-two% were Responders. Within this group, 47% were R12, 28% were R6, and 25% were R2. Among Non-responders, 76% were N6. CF children with meconium ileus (MI) had worse lung function and CXR scores compared to other CF children. Among 53 children with pancreatic insufficiency without MI, R12 had significantly better FEV1 (97-99% predicted) and CXR scores during ages 6-12 years than N6 (89-93% predicted). Both R6 and R2 experienced a decline in FEV1 by ages 10-12 years.
The authors concluded early growth recovery in CF is critical, as malnutrition during infancy tends to persist and catch-up growth after age 2 years is difficult. The longer adequate growth was maintained after early growth recovery, the better the pulmonary outcomes at age 12 year
Don B Sanders (fig.112) heads a research group in the Department of Pediatrics, University of Wisconsin.
Savant AP, McColley SA.Cystic fibrosis year in review 2017. Pediatr Pulmonol. 2018 Sep;53(9):1307-1317. doi: 10.1002/ppul.24081. Epub 2018 Jun 21.[Pubmed]
In this article, the authors highlight cystic fibrosis (CF) reports published in Pediatric Pulmonology during 2017. They also include articles from a variety of journals that are related or are of special interest to clinicians.