Introduction – A history of cystic fibrosis by Dr James Littlewood OBE

“To write an article of any sort is, to some extent, to reveal ourselves. Hence even a medical article is, in a sense, something of an autobiography” – John Chalmers Da Costa (1863-1933). Selected Papers and Speeches, 1931
There follows a miscellany on various aspects of cystic fibrosis (CF) as seen by one person, a general paediatrician from Leeds in the North of England who qualified in medicine in 1956. I was originally a general paediatrician in the true sense of the term involved in all aspects of paediatrics as was usual in the Fifties, but by the nature of the childrens’ problems referred to me following my appointment as a consultant general paediatrician in 1968, I became particularly involved with respiratory, gastrointestinal disorders and neonatal care. Over the next twenty years or so this eventually evolved into an increasing and ultimately a major involvement with the treatment of people with CF and the development of the Leeds Regional Cystic Fibrosis Service .

Jim Littlewood

Daniel Peckham
Since the early Eighties and following my retirement from the National Health Service in 1997, I have had a continual close involvement with the UK Cystic Fibrosis Trust – first as a Member and then Chairman of their Research and Medical Advisory Committee, from 2003 as Chairman of the CF Trust and eventually in 2011 I was honoured to be elected Honorary President
This website is edited and produced by Professor Daniel Peckham, Director of the Leeds Regional Adult CF . Some years ago Daniel offered to put my History on the Leeds CF Website – an offer I quickly accepted and one that has proved very valuable and enjoyable.
HOW TO USE THIS HISTORY
The main format of the History considers the developments by decades, starting each of the earlier decades with a short commentary followed by some of the important references of that decade. One reader referred to the work as a “Paper Trail History” which seems appropriate.
FOR A GENERAL OVERVIEW
– “A Short History of Cystic Fibrosis – from recognition (1938) to treatment of the gene (2012)” is included the end of this introductory section.
– “Introduction” to each decade summarises the progress and significant happenings during that decade.
– Joseph Levy Memorial Lecture – “Looking back over 40 years and what the future holds” delivered at the 2004 International Birmingham CF Conference. The full text of the lecture can be accessed on the internet by entering ” Joseph Levy memorial Lecture” where the full lecture is available .https://www.ecfs.eu/sites/default/files/general-content-files/LEVY_Rossi_lecture.pdf
– Hodson and Geddes’ Cystic Fibrosis Edited by Bush A, Bilton D, Hodson M. FOURTH EDITION (2016). The two chapters below provide a concise account of the history of cystic fibrosis from the early days to the present time.
Appendix A. “History of cystic fibrosis (up to 1989)”. by James M Littlewood
Chapter 1. “Introduction: from the discovery of the CFTR gene in 1989 through to 2014”. by Kris de Boeck.
In the THIRD EDITION (2007) Chapter 1 “The History of Cystic Fibrosis” by James M Littlewood of this textbook is virtually the same as Appendix A of the Fourth Edition mentioned above. This whole chapter can be accessed on the internet by entering – History of Cystic Fibrosis -> scholarly articles ->History of cystic fibrosis -> Littlewood. or by the link https://books.google.co.uk/books?hl=en&lr=&id=9x_cBQAAQBAJ&oi=fnd&pg=PA3&dq=history+of+cystic+fibrosis&ots=TZT97CxqZn&sig=qk1AEtVt3uzPVYvVx0CG8EQ0Fgo#v=onepage&q=history%20of%20cystic%20fibrosis&f=false
FOR A PARTICULAR TOPIC
More frequently discussed subjects (for example antibiotics, sweat tests, neonatal screening, etc ) and the abstracts relating to them mentioned in the main text are also reproduced and classified into their individual sections in the TOPICS section. Click here, where the entries relating to the particular subject appear in chronological order.
The words “above” and “below” after a particular reference indicate the reference or fact is present either earlier or later in the main text; it can be identified there by its date.
FOR AUTHOR IMAGES /INFORMATION
Enter AUTHOR IMAGE INDEX Click here to find an image of an author and brief biographical details. This section can also be accessed directly on line as “Cystic fibrosis authors images”.
Here are listed al the authors whose images are included with their abstract. The date against the individual’s name indicates the date of their entry in the main text where their image and brief biographical details appear. There are more than one entry for some authors. The facility appears to be now widely used by others seeking images of authors of CF articles and there are now more than 1500 authors listed.
FOR SEARCH OF THE WHOLE SITE
Use the “SITE SEARCH” facility to search the whole text for a particular word. When searching for a particular subject it recommended to first look in the appropriate TOPIC section in which subjects are listed in a detailed index.
FOR REFERENCES – EXTRA INFORMATION

Royal Society of Medicine, London
Every reference in the text has a direct link included; either [PubMed] in early years or more recently [pubmed.ncbi.nlm.nih.gov/]
Although the full PubMed abstract is included in many of our summaries are reproduced in full, accessing [PubMed] or [pubmed.ncbi.nlm.nih.gov/] the link is useful as additional previous and more recent references relevant to the particular topic are listed after the particular abstract.
Also a direct link to the full article is present on many of the abstracts.
Although we include PubMed links with many of the earlier references, it was only during the Seventies and Eighties that PubMed started to include an abstract. The information we include on the content of these early references was obtained from my consulting and photocopying the original articles from the bound volumes on shelves of the excellent library of the Royal Society of Medicine in London. This was possible as I stayed frequently overnight in London at the RSM from the early Nineties until 2011 during which time I was first Chair of the CF Trust’s Research and Medical Advisory Committee and Chairman of the CF Trust from 2003 – 2011.
Some publications are designated as “Megapapers”
These are publications that have had a major influence on the understanding, diagnosis, treatment or outlook of people with cystic fibrosis. For example, Dorothy Andersen’s 1938 paper describing the condition (Andersen DH, 1938) following which CF was generally recognised as evidenced by the many reports that soon followed when pathologists reviewed their previous autopsies. Also Paul di Sant’Agnese’s recognition of the abnormal sweat electrolytes (di Sant’Agnese et al, 1953) clearly comes into this category.
Precisely which of many other important publications deserve a “megapaper” rating is a purely personal opinion of a non-scientist clinician, Jim Littlewood, treating hundreds of people with CF and frequently learning from experience and from colleagues whilst doing the job. Some of these articles definitely influenced our treatment of people with CF and others advanced the general understanding of the condition.
Any suggestions of papers that deserve designation as megapapers would be most most welcome.
==================================
SOME DETAILS OF THE DEVELOPMENT OF CF SERVICES IN LEEDS

St James University Hospital, Leeds

Seacroft Hospital, Leeds
The details of the development of the Leeds Regional Paediatric CF service, which started with a small monthly clinic at Seacroft Hospital, Leeds in 1975 and later at St James’s University Hospital, have been described later in this website – (The origins and Development of the Leeds Regional Cystic Fibrosis Unit (Pt1 & Pt2))

Joseph Levy CBE BEM

Ron Tucker OBE
The encouragement and support of the late Mr. Ron Tucker OBE, the then Director of the UK Cystic Fibrosis Trust from 1964, and his frequent advice to parents, patients and doctors in the UK to seek an opinion in Leeds, was a major factor encouraging the early development of our CF service.
Also there was invaluable financial support from the UK Cystic Fibrosis Trust and the Joseph Levy Foundation which allowed us to appoint a CF Research Fellow, Dr Mike Miller (a registrar grade doctor) and CF Nurse Specialist (Mrs. Teresa Robinson) from the early Eighties; both these colleagues, and those who followed them, were absolutely crucial in the development of the CF service and were among the first specialised CF appointments in the UK.

Dr. Mike Miller
Mrs. Teresa Robinson
On this local note, for a service to flourish it must be perceived to be benefiting the patients. This must have been the case as many families and patients returned year after year, often from great distances, even from as far away as Hong Kong, for an Annual Comprehensive Assessments by the small but increasingly expert team at St. James’s University Hospital, Leeds (to where we had moved the 2 miles from Seacroft in 1980)
The Leeds CF Service is recognised by the Yorkshire Regional Health Authority in 1983
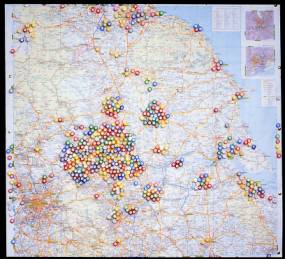
Patients referred to Leeds for Comprehensive CF Assessments
Paediatricians in the Yorkshire Region also found the Comprehensive Assessments of their patients helpful and in 1983 their Regional Paediatric Advisory Committee recommended that the Yorkshire Regional Health Authority officially recognise the service as a “tertiary referral service” for cystic fibrosis – the first to be so recognised and funded in the UK.
The support of the hospital managers over the years was a major factor in establishing the regional status and funding through the Eighties – particularly Mr. Roger Canon, St James’s Finance Manager.

Roger Canon
We are grateful to the numerous colleagues at Seacroft and St James’s Hospitals who, for over 30 years, have contributed their skill and expertise to the care of the patients allowing the development of a CF service which now provides full care for over 500 adults and children with cystic fibrosis (adults at St James University Hospital and children at Leeds General Infirmary); the involvement of these colleagues and their specialist skills was absolutely essential to the development and success of the service.
Access to information on Cystic Fibrosis via modern electronic communications
The major developments in electronic communications in recent years have been timely for an undertaking such as this History. The unprecedented advances in access to previous published work via the Internet, in Medline and PubMed in particular, and the availability and ease of transferring visual images, have presented an opportunity which I hope will add a slightly more human touch to some of the important contributions over the years. To actually see pictures of Harry Shwachman, Paul di Sant’Agnese, Archie Norman and many others, who have made such major contributions to our knowledge of the disorder will I hope add to the interest and value of this account of some aspects of the remarkable story of cystic fibrosis.

Daniel Peckham, Steve Conway, Jim Littlewood and Sister Rachel Metcalfe at opening of the new adult CF unit at St James’s in 2008
These developments resulted in the decision to publish this account on the web so as to provide easy access for all those interested in the story of cystic fibrosis.
The excellent and widely used website developed by Professor Daniel Peckham and his colleagues in Leeds (www.cfmedicine.com and more recently www.cysticfibrosis.online) seemed to be the ideal place to publish this history. I am most grateful for Daniel’s suggestion that we publish it on his Leeds website and for his collaboration, guidance (and patience!) during implementation of the project and since. Daniel has kindly agreed to co-author the New Millennium sections to ensure a CF expert, currently involved in a busy CF centre, would ensure accuracy, relevance and credibility to the content and my personal comments relating to recent developments.
Main sources of information
Where abstracts were available for the more recent papers I have endeavoured to extract the relevant message and reduce the number of statistics as, with the modern electronic databases, the reader can obtain the abstracts or even the full papers if necessary. Also, most of the articles mentioned have a [PubMed] or [pubmed.ncbi.nlm.nih.gov/] link , which from around the mid-Seventies, usually allows access to the full abstract.

Mr. Archie Crompton
I have read and made photocopies of many of the early papers from before 1960 from the originals in the excellent library of the Royal Society of Medicine (RSM) where I stayed regularly during my frequent visits from Leeds to London from 1995 until 2011. During this time I was first Chair of the Research and Medical Advisory Committee and then from 2003 to 2011 Chairman of the Cystic Fibrosis Trust; I had retired from the NHS in 1997. These trips to the RSM library involved many hours in the basement among the old bound journals dating back to the Nineteenth century.
I am also particularly grateful to my friend and erstwhile St James’s surgical colleague Mr. Archie Crompton for the German translations of a significant number of the photocopies of these older articles I recovered from the basement of the Royal Society of Medicine library.

Year Books of Pediatrics from 1934
I worked in the late Professor Stuart Craig’s Leeds University Department of Paediatrics and Child Health from 1960 until moving to the NHS in 1968 as a consultant paediatrician. When he retired in 1967 Professor Craig kindly gave me his US Year Books of Pediatrics dating from 1934 to the late Fifties and these have been an invaluable source of information.

Professor Stuart Craig
The content of the whole of this History is, of necessity, heavily biased towards publications in English, developments in the UK and my own slant on these developments as a working paediatrician. Progress was relatively non-existent during the Forties in the UK and Europe during the Second World War. It is almost certain that over the years many important contributions in German, French and other languages have been overlooked. For this I apologize and do hope that those who recognise these omissions will contact me to rectify the situation. This present on line version of the History can be edited as soon as any omission or error is brought to my attention.
The various phases of research and understanding of Cystic Fibrosis
Some research since the identification of CF in 1938, although of basic scientific interest, was of little or no immediate relevance to the treatment of people with cystic fibrosis. Unfortunately, and understandably, in the past a significant amount of CF research has fallen into this latter category, particularly when there was no clear idea as to the nature of the basic defect. Fortunately, since the early Eighties and particularly after the identification of the CF gene in 1989, much scientific research is clearly more focused on the investigation and correction of the basic defect, by either gene therapy or pharmacological means.

Maurice Super
However, I am in total agreement with the late Dr Maurice Super’s observation that “it is far easier to write on these milestones in retrospect, since discovery of the gene and agreement on the transport defect. For a long time CF was fertile only in the perfusion of false dawns and in controversy between workers who could not reproduce one another’s work. Now we have a fine distillate from what was once very muddy wine” (Super, 1992 below).
Maurice wrote these words in the heady days soon after the identification of the CF gene in 1989 and before the first papers on gene therapy were published from 1993 onwards i.e. before the many problems in correcting the basic defect were experienced. Nonetheless, the identification of the CF gene was the definite turning point in the investigation, understanding and treatment of CF after which there was steady, focused progress in correcting the basic defect by either gene replacement or pharmacological therapy.

Award of the Paul di Sant’Agnese Award in 1989 to the leaders of the successful teams (in italics)- Lap Chi Tsui, Paul di Sant’Agnese, Evelyn and Milton Graub, Francis Collins and Jack Riordan
So cystic fibrosis is truly a disorder of our times

Dorothy Andersen
During the lifetime of many of us, including myself, the condition was first clearly described as a specific entity in 1938 by Dorothy Andersen, a New York pathologist (when I was 6 years old); she also first recognised the heredity aspects in 1946. The treatment has steadily improved, the gene and its product were identified in 1989 and by 2012 the first drug, ivacaftor (Kalydeco), to radically modify the effects of one of the genetic mutations (G551D), became available to treat patients first in the USA; similar very effective modulators became available over the next decade. Also in 2012 the first multi-dose trial of gene therapy commenced by the UK Gene Therapy Consortium and was completed in 2015. Unfortunately, the effect was not sufficient to develop gene therapy which is not yet available in 2023, although it remains a major area of research.
Advances in related fields relevant to cystic fibrosis are discussed
At times I will digress to discuss the impact of parallel developments in related fields of medicine where progress has been central to further understanding and treatment of cystic fibrosis, for such advances have been absolutely essential to the progress in CF research – for example the understanding of the role of gluten in coeliac disease, the many advances in genetics and molecular biology and the many technical advances in investigation and general laboratory medicine
In conclusion
Very many thanks to the numerous colleagues in so many disciplines and many countries who have contributed to our knowledge of CF – not least the patients and their families. These people are too numerous to mention by name. Much of our experience in Leeds has been gained during treatment of hundreds of children and adults with CF – treatment which has been continually changing and improving; the experience gained forms the basis of the “The Leeds Method of Management” which by 2008 had reached the 7th edition; much of the information is now available on this website.
Finally, there will be many people and publications that should have been included but have been omitted and I would welcome comments if there are significant omissions; also if there are factual errors or misinterpretations. Comments from people with CF and their families would also be most welcome. Any anecdotes, images or personal recollections which would add interest to this History would be most welcome.
The text is revised frequently “on line” so there will be an opportunity for immediate alterations and additions rather than waiting for the next edition. The first ten years of the New Millennium were added in late 2010 and with this latest revision we have added 2010 to 2020.
Please do feel free to contact me, Jim Littlewood at jimlittlewood@btopenworld.com
For readers requiring a brief overview there follows one suggestion –
A Short History of Cystic Fibrosis
From recognition (1938) to the first CFTR successful modulator in 2011, to early attempts at gene therapy (2012) and the highly effective modulator in 2018.
Cystic fibrosis (CF) is the most common life-shortening recessively inherited disorder of Caucasian people affecting as many as 1 in 2000 newborn infants in some countries. Both parents are healthy carriers as are up to 1 in 25 of the population. If untreated, signs of severe intestinal malabsorption, failure to thrive and lower respiratory infections, which are slow or fail to clear, are present from an early age. Mutations of the CFTR gene, of which there are now around 2000 described, result in salt and water transport abnormalities across the lining cells of a number of organs leading to viscid secretions which become infected and difficult to clear (lungs) or damage by obstruction (pancreas and liver).
Since 1938, CF has changed from a hopeless condition, fatal in infancy and early childhood, to a chronic condition affecting more adults than children
Thirties

Dorothy Andersen
During the Thirties some infants with so-called “coeliac syndrome” had pancreatic abnormalities noted at autopsy. Dorothy Andersen1, (1) a New York pathologist, reviewed these and her own cases and described them as having “Fibrocystic Disease of the Pancreas”.
Forties
Review of experience at some of the larger children’s hospitals revealed some infants had fibrocystic disease of the pancreas. Sydney Farber recognised CF as a generalised disorder, rather than due entirely to the effects of the malabsorption, and he introduced the term “mucoviscidosis” (2). The recessive mode of inheritance was soon described (3). The major treatment advance in the Forties was the first use of penicillin in 19464 and later other antibiotics (aureomycin and terramycin) (5) without which most of these children did not survive.
Fifties

Paul di Sant’Agnese
Following the heat prostration suffered by some CF infants during the 1948 New York heat wave, Paul di Sant’Agnese later identified the increased salt content of the sweat (6) which became the main diagnostic test. Soon the more convenient pilocarpine iontophoresis was used to stimulate localised sweating (7). There were reports of an improving outlook from the few clinics in N. America gaining experience in treating the condition (8).
In 1957 a young Cleveland paediatrician, Leroy Matthews, funded by the CF parents, introduced a “comprehensive and prophylactic (preventive) treatment programme” which involved early accurate diagnosis, a programme to deal with all the aspects of the disease and data collection to validate the treatment (9, 10, 11).
Physiotherapy, in the form of postural drainage and percussion, the so-called “English” method of physiotherapy12 was introduced and commended by Harry Shwachman (12) – a leading expert for many years.
Sixties
Various lay and professional organisations started. The first, the US National CF Research Foundation (later CF Foundation) had been was formed in 1955 and others followed usually on initiatives of the parents (13).
In 1960, di Sant’Agnese visited Europe and in 1965 the International Cystic Fibrosis (Mucoviscidosis) Association (ICFMA) (now CF Worldwide) was formed in Paris under his chairmanship – the aims to improve the care of people with CF, foster research and increase awareness. The European Working Group for Cystic Fibrosis (Later the European Cystic Fibrosis Society), chaired by Ettore Rossi, soon followed in 1969 (more information can be found here ).
However, although there was modest clinical progress during the Sixties, the nature of basic defect remained totally obscure and there were few scientists researching the problem.
Seventies
During the Seventies there was a definite change to more comprehensive and intensive treatment in a few clinics for example Cleveland whose methods had formed the basis for the network of CF Centres in the USA from 1961 (9,10).
From the early Seventies, Douglas Crozier, who founded the Toronto CF clinic in 1958, abandoned the traditional low fat diet and changed his patients to a high saturated fat diet requiring the patients to take 60-100 pancreatic enzyme capsules daily (15) improving their nutrition, growth and eventually their long term survival (16). In Copenhagen, Niels Hoiby showed by immunological studies that chronic Pseudomonas infection carried a bad prognosis (17) so a policy of regular 3-monthly courses of intravenous antibiotics was introduced for infected patients with an improvement in survival (17, 18, 19).
There was increasing interest in nutrition as survival lengthened. When energy intake was measured accurately, many CF children had a very poor nutritional intake (20). Various regimens and supplements were tried some with modest success, for example an unappetising elemental diet of beef serum protein hydrolysate, glucose polymer and medium chain triglycerides (21, 22). However, the new, acid resistant pancreatic microsphere enzymes (Pancrease and later Creon) became available in the early Eighties and revolutionised the control of the intestinal malabsorption 23.
The few scientists working on CF in the Seventies explored a wide variety of possible abnormalities and serum “CF factors” which may be related to the pathophysiology but by the end of the decade they were no nearer identifying the basic defect.
Eighties

Lap Chi Tsui
This was a decade of remarkable scientific and clinical progress. Michael Knowles demonstration of an abnormally high potential difference across the nasal mucosa providing direct evidence of epithelial dysfunction (24) was followed by Paul Quinton showing by microperfusion experiments that the sweat gland abnormality was chloride impermeability rather than defective exchange (25). Hans Eiberg and co-workers, in Copenhagen, demonstrated a linkage between a liver enzyme marker paraoxinase, which exists in two forms but was present in the same form in 90% of CF siblings (26). In the same year, Lap Chi Tsui demonstrated a marker on chromosome 7 linked to both paraoxinase and cystic fibrosis (27) and in 1989 the CF gene was eventually identified and termed the cystic fibrosis transmembrane conductance regulator (28, 29, 30).
There were many important clinical advances during the Eighties. The CF Foundation’s patient registry and others confirmed a rise in median survival of those patients on the registry from 14 years in 1968 to 20 years in 1977 (31), matched by one centre in the UK (32). The use of the blood immunoreactive trypsin test for neonatal CF screening was a major advance (33) and became the basis of most neonatal CF screening programmes (34,35,36,37). CF Centre care was already well established in some parts of N. America, Europe and Australia where survival improved almost certainly due to CF Centre care (38). Subsequently the UK CF Research Trust gradually financed the appointment of an increasing number of doctors (CF Research Fellows), CF nurses, physiotherapists, dietitians and social workers in the UK hospitals where CF clinics were developing.
During the Eighties, in Europe in contrast to N. America, there was a gradual increase in the use of nebulised anti-Pseudomonal antibiotics to stabilise patients chronically infected with Pseudomonas aeruginosa (39) also to eradicate early P. aeruginosa infection (40, 41) thus delaying or preventing chronic P. aeruginosa infection (42, 43). Intensive courses of intravenous antibiotics became routine treatment for exacerbations of the chest infection and new anti-Pseudomonal antibiotics became available. The repeated courses of intravenous antibiotics required improved venous access and delivery systems e.g. totally implantable venous access devices (44, 45). Also home intravenous antibiotics became available at most CF centres (46, 47, 48).

Magdi Yacoub
A major advance, for those who had reached the end stages of their disease, was heart-lung transplantation in 1985 by Magdi Yacoub in London andJohn Wallwork in Cambridge (49, 50). Bruce Reitz had performed the first successful heart–lung transplant in 1981 on non-CF patient in Stanford; later double lung transplants became more popular (51) and a few surgeons had success with living donor lung transplants (52). The results of liver transplantation in the few patients with severe CF-related liver disease were surprisingly good (53). Various new devices and techniques for physiotherapy of CF were described and evaluated during the Eighties e.g. the forced expiratory technique (54, 55), positive expiratory pressure (56) and high frequency chest compression 57.
Involvement of dietitians and the use of the new acid resistant enzymes improved nutrition (58, 59) – undoubtedly the availability from the early Eighties of the new acid resistant enzymes (23) was one of the major advances in treatment during the Eighties. For the severely malnourished there was nasogastric (60), gastrostomy (61) or parenteral feeding (62). In 1989 the beneficial effect of regular ursodeoxycholic acid treatment in improving CF related liver disease was reported (63). Towards the end of the Eighties more CF Centres for adults were started for the increasing number of people with CF who were now reaching adulthood and the transition of these adolescents to the adult clinics received increasing attention.
Nineties
The steady improvement in clinical progress continued and although the hoped-for gene therapy treatment did not materialise during the decade there was steady scientific progress in this direction. Welsh and his colleagues were the first to achieve correction of the defective chloride channel in CF epithelial cells (64). It was soon shown that the most frequent gene mutation, DF508, was incompletely processed (65) and did not reach the cell membrane (66) but when it did, it functioned reasonably well. Also growing CF cells at a reduced temperature improved expression of CFTR chloride channels (67). Eventually CFTR was confirmed to be an actual chloride channel (68).
It was important to develop an animal model for CF for research to progress and in the early Nineties this was achieved by three groups – in North Carolina (69), Edinburgh (70) and Cambridge (71). Within a year the first report of successful gene transfer into the airways of a CF mouse was reported (72); eventually in 2008 a pig model with CF was produced (73).
In 1993 the first attempt at gene therapy into the nasal passages of a person with CF was reported (74). There followed further reports of gene transfer into animals and humans using either viral or liposome vectors but none resulted in clinically adequate gene expression. However, there were some practical benefits for people with CF resulting from identification of the CF gene including reliable carrier detection, accurate antenatal diagnosis, pre-implantation genetic diagnosis and the incorporation of DNA testing into most neonatal screening programmes.
Attempts to correlate phenotype and genotype indicated that some “mild” mutations were associated with some residual pancreatic function 75. CF carriers were over represented among non-CF infertile men 76, and those with recurrent pancreatitis (77, 78).
The improvement in outlook which characterised the Eighties continued through the Nineties as a result of steady improvement in care and the increasing expertise of the staff at the now well-established CF Centres in many countries. Notable additions to the treatments available included recombinant human DNase (Pulmozyme), the first really effective mucolytic (79,80), a new preparation of tobramycin for inhalation (TOBI) (81) permitting a more widespread use of inhaled antibiotics in N. America, and long term Azithromycin (82) also for patients with chronic Pseudomonas infection. However, the provision of “optimal care”, which is complex and very expensive, for all people with CF proved to be increasingly difficult.
Cross-infection between people with CF, with B. cepacia complex and transmissible P. aeruginosa, resulted in a radical change in the organisation of CF care. In 1977 Pseudomonas (now Burkholderia) cepacia in people with CF was first reported in North America 83 and later in the UK 84; later the organism was shown to spread between patients causing serious even fatal illness (85,86,87,88).
Also there was increasing evidence of cross-infection with particular “highly transmissible” strains of Pseudomonas aeruginosa (89, 90). So the need for cross-infection control radically altered the whole attitude to cross-infection and social contact in both CF Centres and in the community. From the early Nineties all people with CF were strictly segregated according to their microbiological status.
Another significant advance in treatment during the Nineties was the widespread adoption of eradication treatment for early Pseudomonas infection in Europe but not in the USA (41) which subsequently resulted in a significant reduction in the incidence of chronic infection in the UK and Europe (42).
From the early Nineties the new high strength pancreatic enzymes (e.g. Pancrease HL, Creon 25,000) were available and welcomed by people with CF and used by many today although care was needed avoid very high doses which rarely could cause damage to the lower bowel. With increasing survival into adulthood major problems related to CF including diabetes mellitus, liver disease, osteoporosis, pregnancy and infertility became increasingly common and presented further chronic management problems.
The New Millennium
The main scientific research efforts were directed towards better understanding the function and control of CFTR and continuing efforts to treat the basic genetic defect with either gene replacement therapy or pharmacological correctors or potentiators of the mutant gene.
In 1999 three research teams in London, Oxford and Edinburgh were encouraged to form the UK Gene Therapy Consortium (GTC) and eventually a multidose gene therapy trial started in 2013 and successfully completed in 2015 although unfortunately the modest effect was not sufficient to take the product forward (www.cfgenetherapy.org.uk) The use of CF animal models remained an essential part of research, particularly CF mice, which had been available since the early Nineties (69, 70, 71). Eventually towards the end of the decade a CF pig was produced (73) and also a CF ferret (91).
In N. America, and to a lesser extent in Europe, the emphasis of research has been on a variety of pharmacological correctors and potentiators of the abnormal gene (www. cff.org/treatments/pipeline) including gentamicin for “stop mutations” (92) and subsequently of PTC 124 (Ataluren) (93).
The CFTR modulator drugs to treat specific genetic mutations, introduced during the decade following the introduction of the first, ivacaftor, in 2012 (94) undoubtedly represented the most important advance up to that time and radically changed the outlook for the majority of people with cystic fibrosis during the next decade. By 2013 drugs for treating other mutations, including the most frequent Phe508del, were undergoing clinical trials either alone of with Ivacaftor (95). The most recent combination of elexacaftor/tezacaftor/ivacaftor ((known as Trikafta in the USA, Kaftrio in Europe) represents a really major unprecedented advance which has radically changed the outlook for over 90% of patients with one or two Phe508del mutations; the results were quite dramatic. Undoubtedly the treatment of people with CF entered a new era (96,97,98). Unfortunately the drug is very expensive and could be unaffordable in some countries.
What of the Future?
Early diagnosis in the first weeks by neonatal CF screening, early expert advice, support and monitoring by a specialist CF team, and early appropriate treatment of respiratory infection and malabsorption are now well established as essential and should be the right of all people with CF both now and in the future. Chronic gastrointestinal problems of pain and distal intestinal obstruction syndrome are still relatively common and it is encouraging that they are receiving more attention. A greater proportion of care will be provided from Specialist CF Centres – virtually all advances in clinical care have occurred at such centres. The persisting inequalities of care so clearly revealed in the past and yet still present as revealed by the CF registries in North America, the UK and elsewhere, hopefully will lessen.
More user-friendly “conventional” treatment to control symptoms will be available e.g. more efficient nebulisers, powder rather than liquid delivery of inhaled drugs. Also increasing efforts to improve adherence from various psychosocial strategies are of increasing importance.
More lung transplants will be required and measures are being taken to increase the supply of donor organs where this is a problem. Other major and complex problems will increase as the population ages e.g. diabetes, liver disease, osteoporosis, pregnancy, infertility, chronic renal problems, drug allergies as well as complex psychosocial issues. There will be increasing use of home care and CF Nurse Specialists as part of the CF centre team will be increasingly important. Protocols for prevention of cross-infection will be even more important as new organisms emerge and resistance and allergies to current antibiotics increase.
Carrier testing of relatives, the option of antenatal screening and, if positive diagnosis, and pre-implantation diagnosis (PIGD) for high risk couples should be encouraged and available. Unfortunately this opportunity to ensure an infant of two carriers does not have CF by the use of PIGD does not receive the attention it deserves at present.
Major efforts to treat the basic defect evermore efficiently will continue. It is very likely that either gene replacement, into which there is still very active research, or even more impressive pharmacological treatments with the new modulators or both will effectively normalise, or significantly further improve, the disturbed physicochemical condition within the CF airways, so that much less treatment or even no other treatment will be required for the respiratory tract.
There has never been a time when there was more hope of major progress in CF care than the present. In contrast with the really hopeless outlook in the early years, the present state of knowledge, clinical care and the involvement of so many highly regarded scientists would have been beyond belief even 25 years ago. In fairness to the early workers, there have also been massive advances in medicine, science and technology generally, many of which have facilitated the advances in CF research.
While research, prevention, improvement in diagnosis, treatment and provision of optimal care for people at all stages of the condition will remain of the highest priority, progress both in the pharmacological treatment and gene replacement therapy to correct the basic defect is definitely gaining momentum and success in this area is likely to benefit all people with CF even patients whose condition is more advanced.
Jim Littlewood 2023
Based on -“The History of Cystic Fibrosis” by Dr James Littlewood OBE Edited and produced by Prof. Daniel Peckham.
References
1. Andersen DH. Cystic fibrosis of the pancreas and its relation to celiac disease: a clinical and pathological study. Am J Dis Child 1938; 56:344-399.
2. Farber S, Shwachman H, Maddock CL. Pancreatic function and disease in early life. I. Pancreatic enzyme activity and the celiac syndrome. J Clin Invest 1943; 20:827-833.[PubMed]
3. Andersen DH, Hodges RC. Celiac syndrome V. Genetics of cystic fibrosis of the pancreas with consideration of the etiology. Am J Dis Child 1946; 72:62-80.
4. Di Sant’Agnese PA, Andersen DH. Celiac Syndrome IV. Chemotherapy in infections of the respiratory tract associated with cystic fibrosis of the pancreas; observations with penicillin and drugs of the sulphonamide group, with special reference to penicillin aerosol. Am J Dis Child 1946; 72:17-61.
5. Shwachman H, Fekete E, Kulczycki LL, Foley GE. Effect of long term antibiotic therapy in patients with cystic fibrosis of the pancreas. Antibiot Ann 1958-59; 692-699. [PubMed]
6. di Sant’ Agnese PA, Darling RC, Perera GA, Shea E. Abnormal electrolyte composition of the sweat in cystic fibrosis: Clinical significance and relationship to the disease. Pediatrics 1953; 12: 549-563. [PubMed]
7. Gibson LE, Cooke RE. A test for concentration of electrolytes in sweat in cystic fibrosis of the pancreas utilising pilocarpine electrophoresis. Pediatrics 1959; 23:545-549.[PubMed]
8. Shwachman H, Kulczycki LL. Long-term study of 105 cystic fibrosis patients. Am J Dis Child 1958; 96:6-15.[PubMed]
9. Matthews LW, Doershuk CF, Wise M, Eddy G, Nudelman H, Spector S. A therapeutic regimen for patients with cystic fibrosis. J Pediatr 1964; 65:558-575. [PubMed]
10. Doershuk CF, Matthews LW, Tucker AS, Nudelman H, Eddy G, Wise M, Spector S. A 5-year clinical evaluation of a therapeutic program for patients with cystic fibrosis. J Pediatr 1964; 65:677-693. [PubMed]
11. Doershuk CF, Matthews LW, Tucker A, Spector S. Evaluation of a prophylactic and therapeutic program for patients with cystic fibrosis. Pediatrics 1965; 36:675-688. [PubMed]
12. Doyle B. Physical therapy in treatment of cystic fibrosis. Phys Therapy Rev 1959; 39:24 – 27. (Prepared under the direction of Harry Shwachman). [PubMed]
13. Morrison C, Morrison R. National and International Cystic Fibrosis Organisations. In: Dodge JA, Brock DJH, Widdicombe JH, editors. Cystic Fibrosis – Current Topics. Volume 1. England: John Wiley & Sons, 1993:319-346.
14. History of the European Working Group for Cystic Fibrosis and the European Cystic Fibrosis Society Conferences. Based on a lecture by Niels Hoiby adapted by Jim Littlewood (in “Future” section ).
15. Crozier DN. Cystic fibrosis: a not so fatal disease. Pediatr Clin North Am 1974; 21:935-948. [PubMed]
16. Corey M, McLaughlin FJ, Williams M, Levison H. A comparison of survival growth and pulmonary function in patients with cystic fibrosis in Boston and Toronto. J Clin Epidemiol 1988; 41:588-591. [PubMed]
17. Hoiby N. Pseudomonas aeruginosa infection in cystic fibrosis. Diagnostic and prognostic significance of Pseudomonas aeruginosa precipitins determined by means of crossed immunoelectrophoresis. Acta Pathol Microbiol Immunol Scand 1977; Suppl (262): 1-96. [PubMed]
18. Schiotz PO, Hoiby N, Flensborg EW. Cystic fibrosis in Denmark. In: Warwick WJ. Ed: 100 years of Cystic Fibrosis. Minnesota 1981:141-146.
19. Szaff M, Hoiby N, Flensborg EW. Frequent antibiotic therapy improves survival of cystic fibrosis patients with chronic Pseudomonas aeruginosa infection. Acta Paediatr Scand 1983; 72; 651-657. [PubMed]
20. Chase HP, Long MA, Lavin MH. Cystic fibrosis and malnutrition. J Pediatr 1979; 95:337-347. [PubMed]
21. Allan JD, Mason A, Moss AD. Nutritional supplementation in treatment of cystic fibrosis of the pancreas. Am J Dis Child 1973; 126:2-26. [PubMed]
22. Yassa JG, Prosser R, Dodge JA. Effects of an artificial diet on growth of patients with cystic fibrosis. Arch Dis Child 1978; 53:777-783. [PubMed]
23. Khaw KT, Adeniyi-Jones S, Gordon D, Polombo J, Suskind R. Efficacy of pancreatin preparations on fat and nitrogen absorptions in cystic fibrosis. Pediatr Res 1978; 12:437.
24. Knowles MR, Gatzy JT, Boucher RC. Increased bioelectric potential difference across respiratory epithelia in cystic fibrosis. N Eng J Med 1981; 305:1489-1495. [PubMed]
25. Quinton PM. Chloride impermeability in cystic fibrosis. Nature 1983; 301:421-422. [PubMed]
26. Eiberg H, Mohr J, Schmiegelow K, Nielsen LS. Linkage relationships of paraoxinase (PON) with other markers: evidence of PON-cystic fibrosis synteny? Clin Genet 1985; 28:265-271. [PubMed]
27. Tsui L, Buchwald M, Barker D, Braman JC, Knowlton R, Schumm JW et al. Cystic fibrosis locus defined by a genetically linked polymorphic DNA marker. Science 1985; 230:1054-1057. [PubMed]
28. Kerem B-S, Rommens JM, Buchanan JA, Markiewicz D, Cox TK. Chakravarti A et al. Identification of the cystic fibrosis gene: genetic analysis. Science 1989; 245:1073-1080. [PubMed]
29. Rommens JM, Iannuzzi MC, Kerem B-S, Alon N. Rozmahel R. Grzelczak Z et al. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science 1989; 245:1059-1065. [PubMed]
30. Riordan JR, Rommens JM, Kerem B-S, Alon N. Rozmahel R. Grzelczak Z et al. Identification of the cystic fibrosis gene: cloning and characterization of the complementary DNA. Science 1989; 245:1066-1073. [PubMed]
31. 1968 Warwick W, Pogue RE. Computer studies in cystic fibrosis. In Lawson D (ed). Proceedings of 5th international Cystic Fibrosis Conference, Cambridge 1969; 320-330. London. Cystic Fibrosis Research Trust.
32. Mearns MB. Cystic fibrosis. Brit J Hosp Med 1974; Oct: 497-506.
33. Crossley JR, Elliott RB, Smith PA. Dried blood spot screening for cystic fibrosis in the newborn. Lancet 1979; i: 472-474. [PubMed]
34. Heeley AF, Heeley ME, King DN, Kuzemko JA, Walsh MP. Screening for cystic fibrosis by dried blood spot trypsin assay. Arch Dis Child 1982; 57:18-21. [PubMed]
35. Wilcken B, Brown AR, Urwin R, Brown DA. Cystic fibrosis screening by dried blood spot trypsin assay: Results in 75,000 newborn infants. J Pediatr 1983; 102:383-387. [PubMed]
36. Hammond KB, Abman SH, Sokol RJ, Accurso FJ. Efficacy of statewide neonatal screening for cystic fibrosis by assay of trypsinogen concentrations. N Eng J Med 1991; 325:769-774.[PubMed]
37. Farrell PM, Kosorok MR, Rock MJ, Laxova A, Zeng L, Lai HC et al. Early diagnosis of cystic fibrosis through neonatal screening prevents severe malnutrition and improves long-term growth. Pediatrics 2001; 107:1-13. [PubMed]
38. Phelan P, Hey E. Cystic fibrosis mortality in England & Wales and in Victoria Australia. Arch Dis Child 1984; 59:71-83. [PubMed]
39. Hodson ME, Penketh ARL, Batten JC. Aerosol carbenicillin and gentamicin treatment of Pseudomonas aeruginosa in patients with cystic fibrosis. Lancet 1981; i: 1137-1139. [PubMed]
40. Littlewood JM, Miller MG, Ghoneim AT, Ramsden CH. Nebulised colomycin for early Pseudomonas colonisation in cystic fibrosis. Lancet 1985; i: 865. [PubMed]
41. Valerius NH, Koch C, Hoiby N. Prevention of chronic Pseudomonas aeruginosa infection in cystic fibrosis by early treatment. Lancet 1991; 338:725-726.[PubMed]
42. Frederiksen B, Lanng S, Koch C, Hoiby N. Improved survival in the Danish center-treated cystic fibrosis patients: results of aggressive treatment. Pediatr Pulmonol 1996; 21:153-158. [PubMed]
43. Lee TW, Brownlee KG, Denton M, Littlewood JM, Conway SP. Reduction in prevalence of chronic Pseudomonas aeruginosa infection at a regional pediatric cystic fibrosis center. Pediatr Pulmonol 2004; 37:104-110. [PubMed]
44. Cassey J, Ford WD, O’Brien L, Martin AJ. Totally implantable venous access in children with cystic fibrosis. Clin Pediatr 1988; 27:91-95. [PubMed]
45. Essex-Cater A, Gilbert J, Robinson T, Littlewood JM. Totally implantable venous access systems in paediatric practice. Arch Dis Child 1989; 64:119-123. [PubMed]
46. Winter RJ, George RJ, Deacock SJ, Shee CD, Geddes DM. Self-administered home intravenous antibiotic therapy in bronchiectasis and adult cystic fibrosis. Lancet 1984; i: 1338-1339. [PubMed]
47. Gilbert J, Robinson T, Littlewood JM. Home intravenous antibiotic treatment in cystic fibrosis. Arch Dis Child 1988; 63:512-517. [PubMed]
48. Stern RC. Intravenous treatment: where we are and how we got there. In: Cystic fibrosis in the Twentieth Century. C F Doershuk. Editor. Ohio. AM Publishing Ltd, 2001:93-111.
49. Yacoub MH, Banner NR, Khaghani A, FitzGerald M, Madden B, Tsang V, Hodson M. Heart-lung transplantation for cystic fibrosis and subsequent domino heart transplantation. J Heart Transplant 1990; 9:459-467. [PubMed]
50. Penketh A, Higenbottam T, Hakim M, Wallwork J. Heart lung transplantation in patients with end stage lung disease. BMJ 1987; 295:311-314. [PubMed]
51. Pasque MK, Cooper JD, Kaiser LR, Haydock DA, Triantafillou A, Trulock EP. Improved technique for bilateral lung transplantation: rationale and initial clinical experience. Ann Thorac Surg 1990; 49:785-791. [PubMed]
52. Starnes VA, Bowdish ME, Woo MS, Barbers RG, Schenkel FA, Horn MV et al. A decade of living lobar lung transplantation: recipient outcomes. J Thorac Cardiov Surg 2004; 27:114-122. [PubMed]
53. Mieles LA, Orenstein D, Teperman L, Podesta L, Koneru B, Starzl TE. Liver transplant in cystic fibrosis. Lancet 1989; i: 1073. [PubMed]
54. Pryor JA, Webber BA, Hodson ME, Batten JC. Evaluation of the use of forced expiration technique as an adjunct to postural drainage in treatment of cystic fibrosis. BMJ 1979; 2:417-418.[PubMed]
55. Webber BA. Hofmeyer JL, Moran MDL, Hodson ME. Effects of postural drainage incorporating forced expiration technique, on pulmonary function in cystic fibrosis. Br J Dis Chest 1986; 80:353-359. [PubMed]
56. Tyrrell JC, Hiller EJ, Martin J. Face mask physiotherapy in cystic fibrosis. Arch Dis Child 1986; 61:598-611. [PubMed]
57. Warwick WJ, Hansen LG. Long term effects of high frequency chest compression therapy on pulmonary complications of cystic fibrosis. Pediatr Pulmonol 1991; 11:265-271. PMD 1758749
58. MacDonald A. High moderate or low fat diets for cystic fibrosis? In Cystic Fibrosis: Horizons. Ed: Lawson D. 9th International CF Congress, Brighton. Chichester, John Wiley & Sons. 1984; 395.
59. Littlewood JM. An overview of the management of cystic fibrosis. J R Soc Med 1986; 79 (Suppl. 12): 55-63. [PubMed]
60. Bradley JA, Axon AT, Hill GL. Nocturnal elemental diet for retarded growth in a patient with cystic fibrosis. Brit Med J 1979; Jan 20th: 167. [PubMed]
61. Shepherd R, Cooksley WGF, Cooke WD. Improved growth and clinical, nutritional and respiratory changes in response to nutritional therapy in cystic fibrosis. J Pediatr 1980; 7:351-357. [PubMed]
62. Levy LD, Durie PR, Pencharz PB, Corey ML. Effects of long-term nutritional rehabilitation on body composition and clinical status in cystic fibrosis. J Pediatr 1985; 107:225-230. [PubMed]
63. Colombo C, Setchell KD, Podda M, Crosignani A, Roda A, Curcio L et al. Effects of ursodeoxycholic acid therapy for liver disease associated with cystic fibrosis. J Pediatr 1990; 117:482- 489. [PubMed]
64. Rich DP, Anderson MP, Gregory RJ, Cheng SH, Paul S, Jefferson DM et al. Expression of cystic fibrosis transmembrane conductance regulator corrects defective chloride channel regulation in cystic fibrosis airway epithelial cells. Nature 1990; 347:358-363. [PubMed]
65. Cheng SH, Gregory RJ, Marshall J, Paul D, Souza DW, White GA et al. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell 1990; 63:827-834. [PubMed]
66. Kartner N, Augustinas O, Jensen TJ, Naismith Al, Riordan JR. Mislocalization of DF508 CFTR in cystic fibrosis sweat gland. Nat Genet 1992; 1:321-327. [PubMed]
67. Denning GM, Anderson MP, Amara JF, Marshall J, Smith AE, Welsh MJ. Processing of mutant cystic fibrosis conductance regulator is temperature sensitive. Nature 1992; 358:761-764. [PubMed]
68. Bear CE, Li C, Kartner N, Bridges RJ, Jensen TJ, Ramjeesingh M, Riordan JR. Purification and functional reconstitution of the cystic fibrosis transmembrane conductance regulator (CFTR). Cell 1992; 68:809-818.
69. Snouweart JN, Brigman KK, Latour AM, Malouf NN, Boucher RC, Smithies O, et al. An animal model for cystic fibrosis made by gene targeting. Science 1992; 257:1083-1088.[PubMed]
70. Dorin JR, Dickinson P, Alton EWFW, Smith SN, Geddes DM, Stevenson BJ, et al. Cystic fibrosis in the mouse by targeted insertional mutagenesis. Nature 1992; 359:211-215. [PubMed]
71. Ratcliff R, Evans MJ, Cuthbert AW, MacVinish LJ, Foster D, Anderson JR, et al. Production of a severe cystic fibrosis mutation in mice by gene targeting. Nature Genet 1993; 4:35-41. [PubMed]
72. Hyde SC, Gill DR, Higgins CF, Tresize AEO, MacVinish LJ, Cuthbert AW et al. Correction of ion transport defect in cystic fibrosis transgenic mice by gene therapy. Nature 1993; 362:250-255. [PubMed]
73. Rogers CS, Stoltz DA, Meyerholz DK, Ostedgaard LS, Rokhlina T, Taft PJ et al Disruption of the CFTR gene produces a model of cystic fibrosis in newborn pigs. Science 2008; 26; 321(5897):1837-1841. [PubMed]
74. Zabner J, Couture LA, Gregory RJ, Graham SM, Smith AE, Welsh MJ. Adenovirus-mediated gene transfer transiently corrects the chloride transport defect in nasal epithelia of patients with cystic fibrosis. Cell 1993; 75:207-216.[PubMed]
75. Kristidis P, Bozon D, Corey M, Markiewicz D, Rommens J, Tsui LC. Genetic determination of exocrine pancreatic function in cystic fibrosis. Am J Hum Genet 1992; 50:1178-1184.[PubMed]
76. Chillón M, Casals T, Mercier B, Bassas L, Lissens W, Silber S et al. Mutations in the cystic fibrosis gene in patients with congenital absence of the vas deferens. N Engl J Med. 1995; 332:1475-1480.
77. Sharer N, Schwarz M, Malone G, Howarth A, Painter J, Super M et al. Mutations of the cystic fibrosis gene in patients with chronic pancreatitis. N Eng J Med 1998; 339:645-652. [PubMed]
78. Cohn JA, Friedman KJ, Noone PG, Knowles MR, Silverman LM, Jowell PS. Relation between mutations of the cystic fibrosis gene and idiopathic pancreatitis. N Engl J Med 1998; 339: 653-8. [PubMed]
79. Shak S, Capon DJ, Hellmiss R, Marsters SA, Baker CL. Recombinant human DNase 1 reduces the viscosity of cystic fibrosis sputum. Proc Natl Acad Sci 1990; 87:9188-9192. [PubMed]
80. Fuchs HJ, Borowitz DS, Christiansen DH, Morris EM, Nash ML, Ramsey BW et al. Effect of aerosolized recombinant human DNase on exacerbations of respiratory symptoms and on pulmonary function in patients with cystic fibrosis. The Pulmozyme Study Group. N Engl J Med 1994; 331:637-642. [PubMed]
81. Ramsey BW, Pepe MS, Quan JM, Otto KL, Montgomery AB, Williams-Warren J et al. Intermittent administration of inhaled tobramycin in patients with cystic fibrosis. Cystic Fibrosis Inhaled Tobramycin Study Group. N Engl J Med 1999; 340:23-30.[PubMed]
82. Saiman L, Marshall BC, Mayer-Hamblett N, Burns JL, Quittner AL, Cibene DA et al. Macrolide Study Group. Azithromycin in patients with cystic fibrosis chronically infected with Pseudomonas aeruginosa: a randomized controlled trial. JAMA 2003; 290:1749-56. [PubMed]
83. Lararya-Cuasay LR, Lipstein M, Huang NN. Pseudomonas cepacia in the respiratory flora of patients with cystic fibrosis. Pediatr Res 1977; 11:502.
84. Simmonds EJ, Conway SP, Ghoneim AT, Ross H, Littlewood JM. Pseudomonas cepacia: a new pathogen in patients with cystic fibrosis referred to a large centre in the United Kingdom. Arch Dis Child 1990; 65:874-877.[PubMed]
85. Isles A, McLuskey I, Corey M. Pseudomonas cepacia infection in cystic fibrosis: an emerging problem. J Pediatr 1984; 104:206-210.[PubMed]1986
86. Thomassen MJ, Demko CA, Klinger JD, Stern RC. Pseudomonas cepacia colonization among patients with cystic fibrosis. A new opportunist. Am Rev Respir Dis. 1985; 131:791-796.
87. LiPuma JJ, Mortensen JE, Dasen SE, Edlind TD, Schidlow DV, Burns JL et al.
Ribotype analysis of Pseudomonas cepacia from cystic fibrosis treatment centers. J Pediatr 1988; 113:859-862.
88. Govan JR, Brown PH, Maddison J, Doherty CJ, Nelson JW, Dodd M et al. Evidence for transmission of Pseudomonas cepacia by social contact in cystic fibrosis. Lancet 1993; 342:15-19. [PubMed]
89. Cheng K, Smyth RL, Govan JRW, Doherty C, Winstanley C, Denning N et al. Spread of ß-lactam resistant Pseudomonas aeruginosa in a cystic fibrosis clinic. Lancet 1996; 348:639-642.[PubMed]
90. Jones AM, Govan JR, Doherty CJ, Dodd ME, Isalska BJ, Stanbridge TN, Webb AK. Spread of a multiresistant strain of Pseudomonas aeruginosa in an adult cystic fibrosis clinic. Lancet 2001; 358:557-558. [PubMed]
91. Sun X, Sui H, Fisher JT, Yan Z, Liu X, Cho HJ et al. Disease phenotype of a ferret CFTR-knockout model of cystic fibrosis. J Clin Invest 2010; 120:3149-60.
92. Wilschanski M, Famani C, Blau H, Rivlin J, Augarten A, Vital A et al. A pilot study of the effect of gentamicin on nasal potential difference measurements in cystic fibrosis patients carrying stop mutations. Am J Resp Crit Care 2000; 161:860-865.
93. Kerem E, Hirawat S, Armoni S, Yaakov Y, Shoseyov D, Cohen M et al. Effectiveness of PTC 124 treatment for cystic fibrosis caused by nonsense mutations: a prospective phase 11 trial. Lancet 2008; 372:719-727.[PubMed].
94. Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Drevinek P et al. VX08-770-102 Study Group. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. New Engl J Med 2011; 365:1663-72 .[PubMed]
95. Clancy JP,Rowe SM, Accurso FJ, Aitken ML, Amin RS, Ashlock MA et al. Results of a phase IIa study of VX-809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for the F508del-CFTR mutation.
Thorax 2012; 67:12-18. [PubMed]
96 .Keating D, Marigowda G, Burr L, Daines C, Mall MA, McKone EF, Ramsey BW, Rowe SM, Sass LA, Tullis E, McKee CM, Moskowitz SM, Robertson S, Savage J, Simard C, Van Goor F, Waltz D, Xuan F, Young T, Taylor-Cousar JL; VX16-445-001 Study Group. VX-445-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles N Engl J Med. 2018 Oct 25;379(17):1612-1620. doi: 10.1056/NEJMoa1807120. Epub 2018 Oct 18. [Pubmed]
97. Middleton PG, Mall MA, Drevinek P, Lands LC, McKone EF, Polineni D, Ramsay BW, Taylor0Cousar JL, Tullis E, Vermeulen F, marifwada G, Mosowski SM, Nair N, Savage J, Simard C, Tian S, Waltz D, Xuan F, Jain Rassha for the VX17-445-i102 Study Group. Elexacaftor-tezacaftor-iavacaftor for cystic fibrosis with a single The 508del allele. N Eng J Med 2019; 381:1809-19. [Pubmed]
98. Heijerman HGM, McKone EF, Downey DG, Van Braeckel E, Rowe SM, Tullis EMall MA, Welter JJ, Ramsey BW, McKee CM, Marigowda G, Moskowitz SM, Waltz D, Sosnay PR, Simard C, Ahluwalia N, Xuan F, Zhang Y, Taylor-Cousar JL, McCoy KS; VX17-445-103 Trial Group. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: a double-blind, randomised, phase 3 trial. Collaborators (45). Lancet. 2019 Oct 30. pii: S0140-6736(19)32597-8. doi: 10.1016/S0140-6736(19)32597-8. [Epub ahead of print] Full copy available [Pubmed]